Worldwide, cervical cancer is the fourth most common cancer in women and seventh most common cancer overall. In 2012, approximately 528,000 new cervical cancer cases were diagnosed globally. Cervical cancer accounted for 7.5% of all female cancer deaths with approximately 266,000 deaths; the majority (87%) of these deaths occurred in developing countries.1,2 In Europe, the crude incidence of cervical cancer is 13.2/100,000 and the crude mortality rate is 5.9/100,000 women/year;3 in the US, according to the National Cancer Institute (NCI), a total of 12,900 and 4,100 women were estimated to be diagnosed and to have died from cervical cancer in 2015.4
The majority of invasive cervical cancers (70%) are caused by persistent infections with human papilloma virus (HPV) types 16 or 18. This has led to the development of vaccines against HPV16 and 18, which are available and recommended for girls from the age of 9 years, with catchup vaccination for women up to age 26. Although millions of doses have been provided to women and girls, the impact of the vaccine is still decades away.5 Between 80% and 90% of cervical carcinoma are squamous cell carcinomas. The second most common type is adenocarcinomas, which may be pure or mixed (adenosquamous carcinoma). While patients with the adenocarcinoma subtypes may have a poorer prognosis, the treatment recommendations for these subtypes are per the standard of care for cervical cancer as no other therapies have been defined, and these patients are included in all cervical cancer trials.
The Féderation Internationale de Gynécologie et d’Obstétrique (FIGO) cervical cancer staging (stage I–IV) is based on clinical examination, including tumour size (IA, B), vaginal or parametrial involvement (stage IIA–IIIB), bladder or rectum extension (stage IVA) and distant metastases (stage IVB). In its rules for clinical staging, FIGO states that palpation, inspection, colposcopy, endocervical curettage, hysteroscopy, cystoscopy, proctoscopy, intravenous urography and radiographic examination of the lungs and skeleton may be used for clinical staging. FIGO clinical staging criteria do not allow inclusion of computed tomography (CT) scan or magnetic resonance imaging (MRI) for the establishment of stage; however, the current standards of care consider patients with evidence of distant metastasis noted on these imaging modalities as having metastatic or stage IVB disease.
Early-stage cervical cancer is a potentially curable disease by surgery (FIGO stage IA/B1 disease); however, in locally advanced stage disease (FIGO stage II–IVA) the mainstay of primary treatment is combination radiation therapy and radiation sensitising platinum-based chemotherapy. These stages are usually characterised by large central pelvic tumours that are necrotic and often the cause of significant bleeding, as well as the involvement of adjacent pelvic organs (vagina, bladder, rectum, ureters). Up to 70% of patients with bulky primary or advanced disease will have a recurrence, which is generally considered incurable, particularly if distant metastases have developed. Patients with metastatic cancers and those with persistent or recurrent disease after platinum-based chemoradiotherapy not suitable for local control have poor outcomes, with 5-year survival rates between 5% and 15%.6 In this setting any treatment is palliative and the goals of care are to prolong survival but also, and perhaps more importantly, to maintain and/or improve quality of life (QoL).
Chemotherapy, though essentially palliative, is usually recommended for these patients, and although the optimal regimen for chemotherapy has not been defined, cisplatin combination therapy has been considered the standard of care for the last decade. Carboplatin or non platinum regimens (e.g., paclitaxel or topotecan) are options for patients who cannot tolerate cisplatin.5,7 Unfortunately, there are no standard second-line options for these women when their cancer progresses, therefore new therapeutic approaches are urgently required. Conducting clinical trials in cervical cancer patients pose particular difficulties due to their demographic characteristics. Most of the women diagnosed with advanced cervical cancer come from sections of society where, historically, engagement in clinical research has been low and where cost effectiveness and access to new treatments for those in need are major concerns issues.
Targeting angiogenesis is one of the most promising therapeutic strategies to emerge in recent years in the treatment of cervical cancer. Angiogenesis is a critical process in cervical carcinogenesis and tumour progression. Following the publication in 2014 of the randomised phase III study Gynecologic Oncology Group (GOG) 240, the US Food and Drug Administration (FDA) approved the first anti-angiogenic agent, bevacizumab (Avastin©, Genentech/Roche, California, United States), in combination with chemotherapy for use in women with advanced cervical cancer.8 This article will review the rationale for studying antiangiogenic therapy in cervical cancer, focus on the clinical use of bevacizumab and finally highlight potential future directions.
Unmet medical need in advanced cervical cancer and rationale for selection of chemotherapy agents
Improving overall survival (OS) has remained the primary endpoint for clinical trials in advanced cervical cancer, as the prognosis for women with persistent, recurrent or stage IVB cervical cancer remains poor with median durations of OS ≤12 months. In addition, OS has been selected as the primary endpoint because unlike what we see in other cancers, this population is not able to receive multiple lines of chemotherapy.
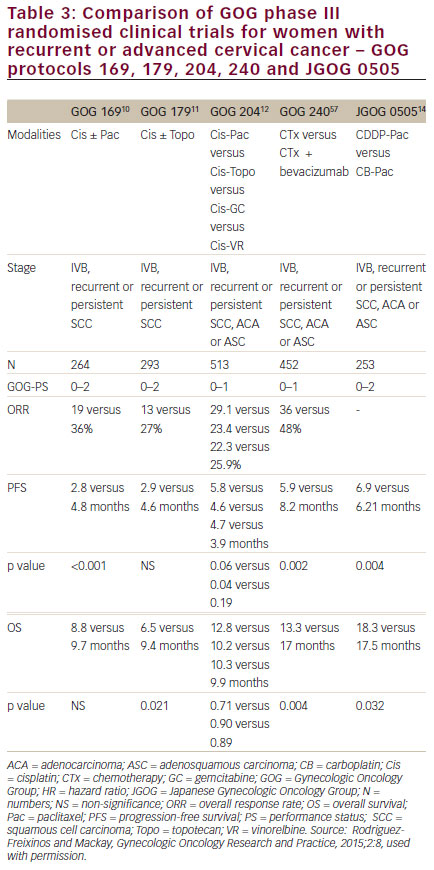
Over the past 3 decades, the GOG has studied the efficacy and tolerability of different cytotoxic regimens for metastatic and recurrent cervical cancer.9The studies GOG-169 and GOG-179 evaluated the clinical efficacy and safety of cisplatin-based doublets against singleagent cisplatin with paclitaxel or topotecan combinations displaying improvements in OS, progression free survival (PFS) and overall response rates (ORR) compared with cisplatin monotherapy. However, only the combination of cisplatin and topotecan achieved a statistically significant impact on ORR, PFS and OS (i.e., difference in median OS of 2.9 months [hazard ratio (HR) = 0.76; 95% confidence interval (CI):
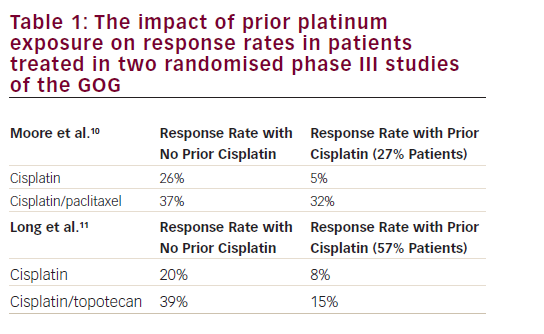
0.593–0.979]).10,11 This study was the basis for the regulatory approval of the combination of topotecan and cisplatin for advanced/recurrent cervical cancer.11
As the use of cisplatin-based chemoradiation had become increasingly prevalent, the GOG analysed the response rates regarding the previous use of cisplatin in studies GOG-169 and GOG-179, only 27% of patients treated on GOG-169 received prior radiosensitising chemotherapy compared with 57% of patients on GOG-179, and noted that for those patients treated previously with cisplatin, the response rates for cisplatin and paclitaxel were superior compared with cisplatin and topotecan (32% versus 15%, respectively) (see Table 1).
These results supported the use of cisplatin and paclitaxel as the reference arm in subsequent GOG studies. In addition, in other words, chemotherapy for patients on GOG-179 was for the most part ‘second-line’ chemotherapy rather than the ‘first-line’ chemotherapy that patients on GOG-169 typically received. The implication is that if tumours have developed acquired resistance to cisplatin at the time of relapse, then the benefit observed in GOG-179 lies primarily with topotecan. Subsequently, study GOG-204, a randomised phase III study, was undertaken in an attempt to establish the best doublet regimen for patients with recurrent and metastatic cervical cancer.12 Study GOG-204 comprised four different platinum-based intravenous doublets containing topotecan, paclitaxel, vinorelbine or gemcitabine. In addition, a health-related QoL analysis through four cycles of therapy and at 9 months follow-up was included. By January 2007, 424 evaluable patients had been enrolled on GOG-204. On 24 April 2007, results of a scheduled interim analysis were presented to the Data Monitoring Committee, indicating that all of the experimental arms on the clinical trial were unlikely to demonstrate improved survival over the control (cisplatin plus paclitaxel) by the end of the study. However, the results observed for OS in the cisplatin/paclitaxel (OS 12.9 months) arm compared with the other three arms (OS 10–10.3 months), shaped the guidelines that support the cisplatin plus paclitaxel combination as the preferred regimen in this disease setting, and as the basis for the standard chemotherapy regimen in the GOG-240 study.5,13
Although the improved toxicity profile conferred by the combination of carboplatin plus paclitaxel clearly has its advantages, the efficacy of this combination for this disease had not been studied within the GOG. In 2006, the Japan Gynecologic Oncology Group (JGOG) opened their phase III study randomised trial to confirm that the regimen carboplatin/ paclitaxel was not inferior to cisplatin/paclitaxel in terms of OS as firstline treatment in patients with advanced cervical cancer. The study reached its primary endpoint since there is no statistical difference between both regimens (HR=0.994; 90% CI: 0.789–1.253 (<1.29), noninferiority one-sided p=0.032 Nevertheless, among patients who had not received prior cisplatin, OS was shorter with carboplatin/paclitaxel (13.0 versus 23.2 months; HR=1.571; 95% CI: 1.06-2.32). Therefore, we may consider carboplatin/paclitaxel as an alternative to current standard of care; however, cisplatin is still the key drug for patients who have not received platinum agents.14
As the cisplatin doublets had been considered the standard of care for advanced/recurrent cervical cancer for the last decades and the majority of patients did not respond, Moore et al. performed a pooled analysis of three published phase III GOG studies of cisplatin versus cisplatin-containing combinations (protocols GOG-110, -169, and -179) to identify factors independently prognostic predictive of response to chemotherapy.15 Four-hundred twenty-eight patients with advanced cervical cancer who received a cisplatin-containing combination in the aforementioned GOG protocols were evaluated. The investigators analysed for baseline clinical characteristics (age, race, performance status (PS), stage, histology, grade, disease site, prior chemotherapy with primary radiation, time to recurrence and single-agent versus combination therapy) and multivariate analysis was conducted to identify factors independently prognostic predictive of response using a logistic regression model. Multivariate analysis identified five factors (African-American ethnicity, PS >0, pelvic disease, prior radiosensitiser, recurrence <1 year) independently predictive of poor response. A simple prognostic index was derived based on the total number of risk factors. When patients were classified into three risk groups (low-risk: 0–1 factor; mid risk: 2–3 factors; high risk: 4–5 factors), patients with 4–5 risk factors were estimated to have a response rate of only 13%, and median PFS and OS of 2.8 months and 5.5 months, respectively. Although this predictive model was externally validated using GOG-149 data that were not used for model development, this model required prospective validation. This prospective validation was undertaken in the phase III GOG-240.
Following the Moore’s prognostic index and due to the poor results shown in the medium- and high-risk group,15 one important question arose. Should those patients still receive treatment based on platinum or should they be treated either with a non-platinum combination regimen or be enrolled into clinical trials with novel drugs? Therefore, it became important that non-platinum doublet therapies underwent rigorous investigation in the recurrent/metastatic setting. Two studies addressed this matter. Symonds et al. reported their results of a Scottish phase II trial of docetaxel (75 mg/m2 day 1) plus gemcitabine (1,000 mg/m2 days)1,8 as second-line chemotherapy in cervical cancer (SCOTCERV study).16 Among 24 evaluable patients, 23 had prior chemoradiotherapy. The principal toxicity was neutropaenia (grade 3 in eight patients, grade 4 in eight patients), with four patients experiencing grade 3 febrile neutropaenia. Dose reductions occurred in 29% of patients (docetaxel) and in 25% of patients (gemcitabine). Importantly, haematological toxicity resulted in the day-8 gemcitabine dose being omitted in 41% of cycles. Among 18 patients evaluable for response, there was one complete response (CR), four partial responses (PRs), six stable disease (SD), and seven progressive disease (PD). The investigators acknowledged that although this combination exhibited some activity against ‘platinum-resistant’ metastatic cervical cancer, the ability to deliver day-8 gemcitabine was compromised. Tiersten et al. published the second study.17Since topotecan and paclitaxel have shown activity alone and in combination with cisplatin in metastatic cervical cancer, the non-platinum doublet of topotecan plus paclitaxel may be suitable for study in this population of potentially platinum-resistant patients. Tiersten et al. analysed the combination of paclitaxel plus topotecan in 15 patients with recurrent, persistent or metastatic cervical carcinoma. Fourteen had received prior pelvic irradiation. Patients were treated with paclitaxel 175 mg/m2 on day 1 and topotecan 1 mg/m2 on days 1–5 of a 21-day cycle with growth factor support. Among 13 evaluable patients, there were 7 (54%) responses (one CR; six PR), and three patients (23%) experienced SD. The PFS and OS were 3.77 and 8.62 months, respectively. Grade 3/4 toxicities included anaemia (47%), leukopenia (27%), thrombocytopenia (13%), neurotoxicity (13%) and diarrhoea (13%). These results led to the consideration of paclitaxel/topotecan regimen as comparator arm in the GOG-240 study.
Angiogenesis in cervical cancer and rationale for targeting
Angiogenesis
Angiogenesis is the development of new blood vessels in areas of new tissue growth. This is a normal physiological phenomenon associated with wound healing and embryogenesis. It is also an important process that occurs almost universally in solid tumours as the result of the enlarging cancer mass and its subsequent growth away from the existing blood supply.
Angiogenesis is a highly ordered process that involves the regulation of multiple signalling pathways and requires interaction between different cell types namely, endothelial cells, stromal cells (fibroblasts) and their interaction with the extracellular matrix, cytokines and growth factors, which leads to the effective formation of new blood vessels. An important interplay of pro-angiogenic signalling occurs in response to the hypoxic state. A protein called hypoxia-inducible factor (HIF)-1 alpha is stabilised in these conditions and enters the nucleus where it forms a complex with another protein (HIF-1 beta). This complex is then able to act as a transcription factor allowing upregulation of growth factors including vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR)-2.18 Although numerous proangiogenic factors have been described there is universal agreement that the VEGF family of ligands, (VEGF-A to –D) and placental growth factor (PLGF) and their associated receptor tyrosine kinases (VEGFR)-1, 2 and 3 are the most important regulators of angiogenesis. VEGF-A, usually referred to as VEGF, binds to VEGFR-1 and VEGFR-2; the stimulation of endothelial cell mitogenesis and vascular permeability is mediated by its interaction with VEGFR-2.19 PLGF and VEGF-B selectively bind to VEGFR-1 and stimulate vessel growth and maturation and recruit pro-angiogenic bone marrow-derived progenitors.20,21 VEGF-C and VEGF-D primarily interact with VEGFR-3 stimulating lymphangiogenesis.22 Other crucial steps in physiological angiogenesis involve the recruitment of pericytes. Pericytes, recruited primarily by platelet-derived growth factor (PDGF), secreted by endothelial cells, are essential for the stabilisation, maturation and support of new vessels.23
Many cancers exploit aberrant angiogenic mechanisms to stimulate tumour growth and metastasis. Tumour angiogenesis was established as a potentially attractive therapeutic target for the treatment of cancer with the publication of Folkman’s hypothesis in 1971.24 Angiogenesis is required for tumour growth beyond 1–2 mm3, when the tumour demand for oxygen and nutrients surpasses the local supply and the hypoxic microenvironment, through the expression of HIFs leads to the activation of angiogenesis.
Tumour-related angiogenesis, in contrast to physiological angiogenesis, leads to a more disorganised vasculature, which is also more permeable,limiting the delivery of drugs to tumour cells. Anti-angiogenic agents have been shown to transiently ‘normalise’ the tumour vasculature, resulting in an increased delivery of oxygen and drugs into the tumour microenvironment.25
Greater understanding of these pathways continues to provide valuable insights into the molecular mechanisms that underlie tumour angiogenesis and provide a foundation for the development of novel anti-angiogenic therapeutic strategies.
Angiogenesis in cervical cancer
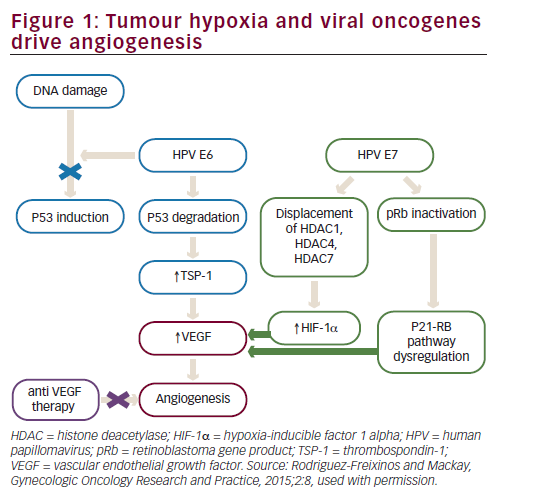
High-risk HPV subtypes 16 and 18 (although other subtypes have also been implicated) are responsible for approximately 70% of invasive cervical cancers.26 Emerging data suggest that viral integration into the host cell genome results in overexpression of a number of host genes, which are potential drivers of carcinogenesis.27 However, the HPV oncoproteins E5, E6 and E7 are the primary viral factors responsible for initiation and progression of cervical cancer. E6, E7 and, to a lesser extent, E5 play important roles in upregulating angiogenesis through the VEGF pathway through their effects on p53 degradation, HIF-1α and inactivation of retinoblastoma protein (pRb). HPV E6 promotes p53 ubiquitination and degradation after E6-p53 binding. Degradation of p53 promotes angiogenesis by downregulating thrombospondin-1 and by increased production of VEGF. HPV E7 results in abrogation of pRb function resulting in p21-RB pathway dysregulation thereby increasing VEGF. In addition, HPV E6 (in a p53 independent manner) and E7 also enhance the induction of HIF-1α, thus increasing VEGF through a second mechanism (see Figure 1).28–31
Over the past decade, the relationship between HPV-16- and 18-associated cervical tumours, hypoxia, markers of tumour angiogenesis and prognosis has emerged. Initial descriptions of high-risk HPV related premalignant cervical lesions seen on colposcopy included atypical angiogenic proliferation along the basement membrane and suggested a role for angiogenesis in the transition from premalignant lesions to invasive cervical carcinoma. Microvessel density (MVD) has been reported to increase with the grade of pre-malignant lesions.32
Tumour angiogenesis (as reflected by the MVD) has been studied as prognostic factor in cervical cancer. Cooper et al. evaluated formalinfixed paraffin-embedded tumour biopsies in 111 patients treated with pelvic irradiation for MVD.33 On multivariate analysis, MVD was a significant prognostic marker, as high tumour vascularity was associated with lower OS and locoregional control. However, these data are controversial. Almost a decade later a prospective analysis of various angiogenesis markers utilising immunohistochemistry was performed on cervical tumour specimens from a multicentre randomised phase III trial (Southwest Oncology Group 8797/GOG-109/ Radiation Therapy Oncology Group 91-12).34 This study investigated the significance of adding cisplatin-based chemotherapy to adjuvant radiation following radical hysterectomy when high-risk factors, such as nodal metastases were present. In contrast to the prior study, the investigators showed that increased tumour angiogenesis (as measured by CD31-MVD [a non-specific endothelial marker] was an independent prognostic marker for PFS and OS [HR=0.36, 95% CI: 0.17–0.79; p=0.010]). In an ad hoc analysis of GOG-109,35 specimens were assessed for expression of markers of tumour angiogenesis including VEGF, thrombospondin (TSP)-1 (anti-angiogenesis factor) and CD105 (tumour-specific endothelial marker). In contrast to CD31 results, the presence of CD105-positive vessels in cervical cancer samples has
shown an association with risk of lymph node metastasis, and worse PFS and OS.36 The differences in outcome observed in these studies may be related to the method used to study MVD. Some markers such as CD31, used in GOG-109, may reflect ‘good angiogenesis’, with CD31- positive endothelial cells exhibiting organised vasculature, potentially leading to well-vascularised and oxygenated tumours, leading to better outcomes, while other markers such as CD105 may indicate a more disordered endothelial structure resulting in poorer outcomes. In addition, markers of tumour angiogenesis can also be evaluated in cervical cancer patients through serological testing. Analysis of VEGF has shown increased VEGF expression in cervical intraepithelial neoplasia grade 3 and squamous cell carcinoma compared with control cervical tissue. In the cervical cancer samples higher VEGF levels were associated with advanced stage disease, increase risk of nodal metastasis and worse PFS and OS.37 In cervical carcinomas, elevated serum VEGF has been identified as a poor prognostic factor.38,39
Angiogenesis plays a pivotal role, not only in initiation of cervical cancer, but also in proliferation and progression of the disease, hence targeting angiogenesis has emerged as a rational therapeutic approach.
Rationale for bevacizumab therapy in advanced and recurrent cervical cancer – The GOG-240 – a shift in the standard of care
The inability of conventional cytotoxic agents to enhance long term survival is likely multifactorial. Since radiation has been the primary treatment modality for locally advanced cervical cancer, women suffering from metastatic (stage IVB) and/or recurrent/persistent cervical cancer typically have been previously irradiated and therefore harbour radioresistant and chemoresistant tumour cell populations where the mechanisms of drug resistance overlap. These patients may have one or more of the following: (1) recurrent tumours within the irradiated field and therefore devascularised areas of poorly healing tissue that are difficult to bathe in chemotherapy, (2) nephropathy as a consequence of a blocked kidney (limiting the ability to clear cytotoxic compounds from the bloodstream) or (3) be at risk for myelosuppression upon exposure to cytotoxic therapy due to depleted bone marrow following prior pelvic radiation. As we have summarised
above we already have enough evidence that supports the relevant role of angiogenesis in cervical cancer. Therefore, targeting angiogenesis seems to be a promising approach in the treatment of advanced or recurrent cervical cancer.
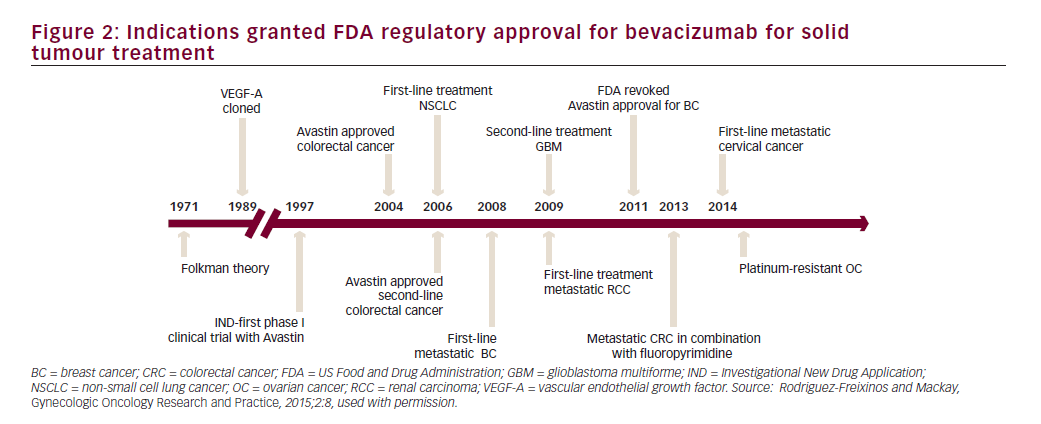
Bevacizumab is a recombinant humanised monoclonal immunoglobulin (Ig)-G1 antibody directed against VEGF-A, which blocks signal transduction through VEGFR-1- and 2-associated pathways. In preclinical models, bevacizumab suppressed VEGF-induced tumour growth and reduced tumour MVD. Bevacizumab appeared to normalise primitive tumour vasculature, leading to an increase in tumour oxygenation and potentially enhancing delivery of cytotoxic agents thereby potentiating their efficacy.40 Bevacizumab has shown clinical activity in different solid tumour types resulting in approval by the FDA for treatment of metastatic colorectal cancer, non-small cell lung cancer, renal cell carcinoma, glioblastoma multiforme and ovarian cancer (see Figure 2).
Wright et al. provided data supporting the use of bevacizumab in combination with chemotherapy in the metastatic cervical cancer setting. A retrospective analysis of women with recurrent cervical cancer was performed showing a meaningful clinical benefit rate of 67% in a heavily pretreated patient population (median of three prior regimens), and acceptable safety when bevacizumab was given at 15 mg/kg intravenous every 3 weeks in combination with cytotoxic chemotherapy.41 Moreover, in a phase II GOG trial of single agent bevacizumab (GOG-227C) in patients with persistent or recurrent squamous cell carcinoma of the cervix and one or two prior cytotoxic regimens (not including prior cisplatin-based chemotherapy concomitantly administered with primary pelvic radiation), 24% of patients survived progression free for at least 6 months, 11% of patients had PRs, the median response duration was 6.21 months (range: 2.83– 8.28 months) and the median PFS and OS of 3.4 months (95% CI: 2.53– 4.53 months) and 7.3 months (95% CI, 6.11–10.41 months), respectively.42 These data compared favourably with historical single-agent cytotoxic phase II studies in similar previously treated patient populations and provided supportive evidence of the activity of single agent bevacizumab in the second and third line treatment of patients with recurrent cervical cancer. Despite these encouraging results, none of the trials in recurrent or metastatic cervical cancer over the last decade have resulted in a significant increase in OS (with median durations of OS ≤12 months) and remains a high unmet medical need. With the rapid incorporation of cisplatin-based chemoradiation therapy over the past 15 years, the concern for platinum resistance in recurrent or persistent tumours has become an important factor, leading to the mandatory development of new therapeutic agents since the standard cisplatin doublets regimens have shown to have minimal impact on survival.
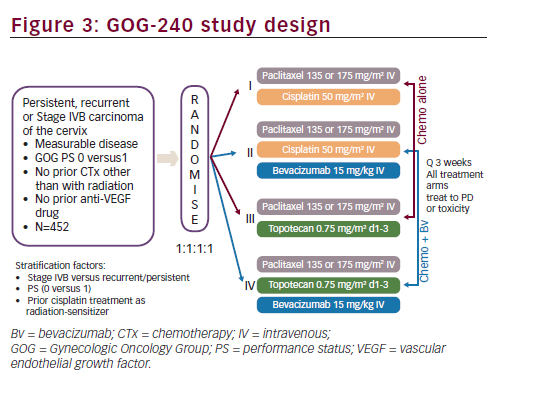
Based on the encouraging results seen in GOG-227C study, a four-arm prospective, randomised clinical trial, the GOG-240, was conducted. The GOG-240 study sought to investigate two important questions for the treatment of recurrent or metastatic cervical cancer: 1) whether outcomes could be improved with the addition of bevacizumab to chemotherapy; and 2) whether outcomes could be improved with the non platinum doublet of paclitaxel plus topotecan. The GOG- 240 study, in order to test these two hypothesis in a limited patient population, used a 2 x 2 factorial design. A major advantage in selecting a 2 x 2 factorial design is the ability to consider the effects of more than one factor at the same time.43 As clinical trials in cervical cancer may typically occur over many years and may require large numbers of patients in a population with limited numbers of patients, this study design allows study of important clinical questions with fewer patients in a shorter period of time. The efficiency and validity of the 2 x 2 factorial design depends upon the absence of interaction between two factors or treatments being studied so that the effects of both factors on the primary efficacy variables follow an additive model. The GOG-240 clinical trial was designed with the assumption that there was no interaction between bevacizumab and the selected chemotherapy backbones (cisplatin, topotecan and paclitaxel) since bevacizumab and these chemotherapeutic agents do not have related mechanisms of action. Study GOG-240 enrolled women ≥18 years of age with primary stage IVB, recurrent or persistent squamous cell carcinoma, adenosquamous carcinoma or adenocarcinoma of the cervix, which was not amenable to curative treatment with surgery and/or radiation therapy. Patients treated on study GOG-240 could be defined as stage IVB based on metastatic disease noted on imaging modalities other than chest X ray (e.g., CT/MRI) and do not, therefore, fall strictly within the FIGO staging criteria. All patients had measureable disease and had at least one ‘target lesion’ to be used to assess response as defined by GOG Response Evaluation Criteria in Solid Tumors (RECIST) criteria. Patients were required to have a GOG PS of 0 or 1. Per the 2 x 2 factorial design of the study, eligible patients were randomly assigned in a 1:1:1:1 ratio to receive one of four treatment arms (see Figure 3). Stratification factors included stage IVB versus recurrent/persistent disease, PS 0-1 and prior concomitant cisplatin and radiation. In all treatment regimens, cycles were repeated every three weeks, and patients continued on study treatment until disease progression or unacceptable toxicities prohibited further therapy. If patients experienced a CR on study they could receive an additional two cycles of therapy at their physician’s discretion and then discontinue study therapy.
The GOG-240 study was activated on 9 April 2009, reaching target accrual on 2 January 2012, for a total of 452 patients. The primary efficacy endpoint was OS: to determine whether the addition of bevacizumab to chemotherapy improves OS and whether the regimen of paclitaxel and topotecan (non platinum) improves OS in comparison with the standard cisplatin and paclitaxel regimen, both objectives in patients with persistent, recurrent or stage IVB carcinoma of the cervix. In addition, the study also had as primary endpoint, to determine and compare the frequency and severity of adverse events (AEs) as assessed by NCI Common Terminology Criteria for Adverse Events (CTCAE) Version 3.0 for the regimens administered in this study. Secondary endpoints in this study were the following: PFS from date of randomisation; ORR (GOG RECIST) and health-related QoL (HRQoL) as measured by the Functional Assessment of Cancer Therapy-Cervix (FACT-Cx), the Functional Assessment of Cancer Therapy for patients with neurotoxicity questionnaire (FACT/GOG-Ntx4) and pain as measured by the Brief Pain Inventory (BPI)-‘worst pain’ single item. Moreover, GOG-240 addressed the important following exploratory efficacy objectives. The study wanted to validate the Moore model, i.e. prognostic markers for predictive modelling. The effect of smoking on prognosis and exploratory and translational research objectives were also evaluated; however, these aforementioned analyses have not been completed and the results are not included herein. Sample size calculation was based on increasing the median OS from 12 to 16 months, detecting with 90% power and a reduction in the risk of death of at least 30%, with the one-sided type I error rate limited to 2.5% for each regimen.
The intent-to-treat (ITT) population comprised all 452 patients randomised to study treatment (225 in the chemotherapy alone group; 227 in the bevacizumab containing groups). Patients were grouped according to which treatment they were randomised to. Twelve patients (five in the chemotherapy group; seven in the bevacizumab-containing group) did not receive any protocol study treatment, and were excluded from the safety population. Hence, the safety analyses were based on 440 randomised patients who received at least one full or partial dose of any component of the study treatment (bevacizumab or chemotherapy) during the study period. Patients were treated at a total of 159 sites (434 patients) in the US and six sites in Spain (18 patients), with a median of two patients per site (range: 1–15). At the time of the clinical cutoff date (12 December 2012), 217 patients (96.4%) in the chemotherapy alone group and 206 patients (90.7%) in the bevacizumab-containing group had discontinued study treatment. The majority of patients in both groups discontinued study treatment because of disease progression relapse during active treatment; a greater proportion of these were in the chemotherapy group (chemotherapy alone: 51.6% versus chemotherapy + bevacizumab: 38.3%).
The study population consisted of 452 adult women; overall, baseline disease characteristics were well balanced between the chemotherapy alone and bevacizumab containing groups (see Table 2). Median age was 46.0 (range: 20–83) years. The majority of patients were ≤65 years of age and white and, notably, had a PS of 0 (chemotherapy alone: 58.7% versus chemotherapy + bevacizumab: 57.7%). Most of the patients in both groups had squamous cell carcinoma (chemotherapy alone:
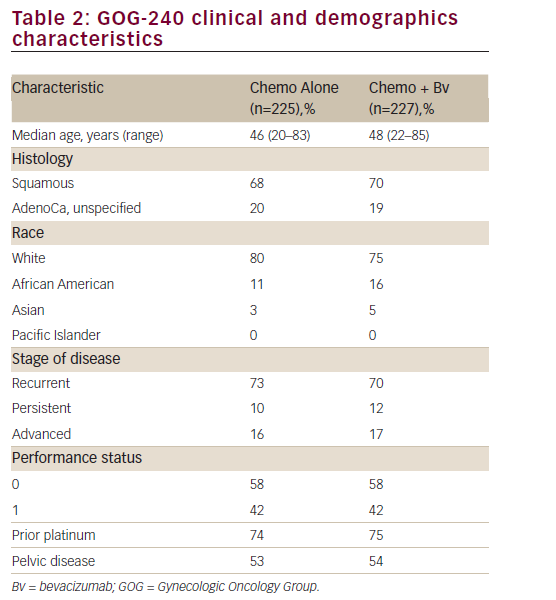
67.1% versus chemotherapy + bevacizumab: 69.6%) and persistent/ recurrent disease (chemotherapy alone:83.6% versus chemotherapy + bevacizumab: 82.8%). Only 16% of patients in each arm presented with advanced disease at diagnosis. The proportion of prior platinum chemotherapy in combination with radiotherapy (only prior radiation sensitising chemotherapy was allowed) was also well-balanced between each arm (74% and 75% in the chemotherapy and the investigational arm, respectively; p=0.666). Furthermore, the majority of patients had a platinum-free interval ≥ 6 months (chemotherapy alone: 72.5% versus chemotherapy + bevacizumab: 64.4%). Approximately 80% of patients had received prior radiation therapy and no patient had received prior non-protocol biological response modifiers.
A pre-planned interim analysis after 174 deaths to determine futility/ superiority was conducted on 6 February 2012 and presented at the Society of Gynecologic Oncology (SGO) meeting in 2013.44 At the time
of this interim analysis (the first data freeze), 62% of patients were alive, with a median follow-up of 12.5 months. Since the majority of patients were not platinum-naïve it was reasonable to expect the non-platinum doublet to outperform the cisplatin-paclitaxel doublet, but the analysis did not find this to be the case. The analysis showed that the topotecanpaclitaxel arm was not superior or inferior to the cisplatin-paclitaxel arm (median OS 15 versus 12.5 months, respectively, HR=1.20; 95% CI: 0.82–1.76).
Essentially, we learned that retreatment with platinum can still be active. Even though these patients received platinum once, we found that that did not preclude it from working a second time. It is important to point out that although the non-platinum doublet was not superior to cisplatinpaclitaxel, it did not underperform either (i.e. it was not inferior). If the non-platinum doublet was significantly underperforming it may have made it harder to interpret an OS advantage with the bevacizumab-containing regimens because half of the patients on this study who received bevacizumab received it in conjunction with the non-platinum doublet.
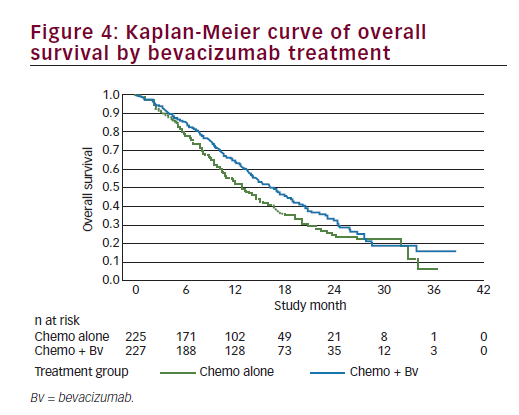
The efficacy analysis for OS was performed using the clinical cutoff date (12 December 2012) with 271 OS events. Study GOG-240 met its primary endpoint by demonstrating improved OS in patients treated with bevacizumab in combination with chemotherapy compared with chemotherapy alone (HR=0.71; 95% CI: 0.54–0.95; p=0.0035) with an improvement in the median OS of 3.7 months (from 13.3 months with chemotherapy alone to 17 months with chemotherapy + bevacizumab). We should note that the Kaplan-Meier plot showed initial separation of the curves in favour of the chemotherapy + bevacizumab group at approximately 3 months after randomisation and they remained separated until approximately 28 months at which time there were few patients remaining at risk (see Figure 4). In addition, there was no statistical evidence of differences in OS between the two chemotherapy backbones (HR=1.15; 95% CI: 0.91–1.46; log-rank p=0.2326), with a median duration of OS of 15.5 months in the ciplatin + paclitaxel ± bevacizumab group and 13.3 months in the topotecan + paclitaxel ± bevacizumab group); the study showed that topotecan in combination with paclitaxel and bevacizumab provided a clinically meaningful benefit and is an acceptable alternative to cisplatin chemotherapy. Furthermore, the analyses of OS by individual trial treatment arms support the conclusions of the primary pooled analyses. OS improved by a median of 3.2 months when bevacizumab was used in combination with cisplatin + paclitaxel (14.3 months to 17.5 months; p=0.03) and by a median of 3 months when bevacizumab was used in combination with topotecan + paclitaxel (12.7 months to 16.2 months; p=0.08) and, therefore, the addition of bevacizumab to either chemotherapy backbone improved OS. The consistency of OS results was examined across subgroups defined by patient demographic and baseline disease characteristics. Subgroup analyses for OS were, in general, consistent with the overall analysis of OS for all patients. In nearly all subgroups the HRs were less than or equal to 1, indicating a benefit in OS for the chemotherapy + bevacizumab group in comparison with the chemotherapy alone group. This fact is even more relevant among patients with prior platinum exposure or with recurrent disease in the pelvis after prior radiation. A point of contention is the results in the histology subgroups. In the two subgroups of adenocarcinoma and adenosquamous carcinomas, the HRs were 1.17 and 1.20, respectively; however, the 95% CIs were wide, probably due to the small sample size. Though there has been some suggestion that adenocarcinoma subtypes of cervical cancer have a poorer prognosis, there are currently no data that define different treatment measures for this patient group and these patients are treated per the standard of care for cervical cancer. The results from GOG-240 provide supportive evidence to suggest that any therapy that improves outcomes should be considered applicable regardless of histological subtypeuntil definitive data to the contrary are available.
The improvement in the primary efficacy endpoint of OS was supported by the positive results in PFS for patients with persistent, recurrent or stage IVB carcinoma of the cervix. The addition of bevacizumab to chemotherapy led to a clinically meaningful and statistically significant increase in PFS (HR=0.67; 95% CI: 0.54–0.82; p=0.0002) and an improvement of 2.3 months in the median time to progression or death (8.2 months in the chemotherapy + bevacizumab group compared with 5.9 months in the chemotherapy alone group). The Kaplan-Meier plot of PFS showed separation of the curves approximately 2 months after randomisation in favour of the chemotherapy + bevacizumab group, which was maintained over time until approximately 24 months when there were few patients at risk.
A statistically significant and clinically meaningful improvement in ORR (based on GOG RECIST) was also observed in patients who received bevacizumab in combination with chemotherapy compared with chemotherapy alone, 48% versus 36% respectively (p=0.008). Interestingly, of the 452 patients enrolled on study, there were 44 (9.7%) complete responders (28 in the chemotherapy/bevacizumab arm and 16 in the chemotherapy arm) defined as a complete regression of all target lesions over an 8-week period. Recently, a post-trial ad hoc analysis was presented as abstract form only in the SGO Meeting 2015. The objectives of the ancillary data study were to identify clinical and/or pathological factors associated with CR on GOG-240, as well as possible predictors of response to anti-angiogenic therapy. Patients achieving a CR in both treatment arms (chemotherapy alone and chemotherapy + bevacizumab) were well balanced in terms of age, PS, histology, FIGO stage, baseline tumour size and prior platinum and radiation therapy. The majority had squamous histology (n=31; 70%), recurrent/persistent disease (n=39; 81%) and had been previously irradiated (n=37; 84%). Remarkably, 18 patients (41%) achieved CR in the irradiated field, most of whom received bevacizumab treatment (n=11; 61%). CR is associated with prolonged OS. Median PFS and OS were 18.3 and 39.3 months, respectively. Median OS for patients with CR on the cisplatinpaclitaxel- bevacizumab arm has not been reached.45
As we have shown previously, GOG-240 met one of its primary endpoints with the regimens administering the anti-VEGF monoclonal antibody, bevacizumab, being associated with significantly improved OS. A major objective of GOG-240 was to prospectively validate the Moore criteria and apply it to the group of patients treated with chemotherapy plus bevacizumab to try to identify a cohort of patients that were not likely to benefit from anti-angiogenesis therapy. The Moore criteria themselves were highly prognostic for OS, PFS, and ORR, across the low-, mid-, and high-risk subgroup stratification. Patients on the lowrisk cohort experience 21.8 months OS and 57% RR, while those in the high-risk cohort have significantly worse OS of 8.2 months (p<0.0001) and 18.5% RR (p<0.001). In addition, high-risk patients as defined by Moore´s prognostic index appeared to derive the greatest OS benefit from experimental treatment arms with bevacizumab compared with chemotherapy alone. The median OS was 6.3 months versus 12.1 months, respectively (HR=0.377). Conversely, low-risk patients may not experience significant survival gains from the addition of bevacizumab to chemotherapy compared with chemotherapy alone (21.8 months versus 23.0 months, respectively, HR=1.119). Taking into account these results, the burning question is: can the Moore Criteria be used to identify subgroups of patients whose prognosis is so poor that they are unlikely to benefit from anti-VEGF therapy? Surprisingly, bevacizumab appears to reduce the hazard of death to a greater extent in patients with a high-risk score, while the benefit of bevacizumab in low-risk patients is unclear.
Overall, the safety data observed in the GOG-240 study demonstrated an AE profile consistent with that seen with bevacizumab treatment across indications; however, the incidence of gastrointestinal vaginal fistulae, such as rectovaginal fistulae, was higher in this study in bevacizumab treated patients (chemotherapy alone: 0.9% versus chemotherapy + bevacizumab: 8.2%) than previously observed. This may be due in part to the patient population in GOG-240 study: patients studied had a significantly higher rate of prior pelvic radiation therapy compared with abdominal and pelvic radiation in other indications studied and this may be a pre-disposing factor to the development of such fistulae. Actually, fistulae appeared to occur exclusively in patients who had undergone prior pelvic radiotherapy.47 As expected, Adverse Events of Special Interest (AESIs) based on NCI CTCAE grade 1 to 5 were observed at a higher percentage in the chemotherapy + bevacizumab group (39.9%) compared with the non-bevacizumab containing groups (16.7%), of which most were grade ≥3 (chemotherapy alone: 16.7% versus chemotherapy + bevacizumab: 37.6%). This difference was driven by an increase (≥5%) in the incidence in the bevacizumab-containing groups relative to the chemotherapy alone group of the following events: hypertension (chemotherapy alone: 0.5% versus chemotherapy + bevacizumab: 11.4%). Nevertheless, no patients in GOG-240 were taken off study for treatment-induced hypertension; gastrointestinal perforations (chemotherapy alone: 0.5% versus chemotherapy + bevacizumab: 10.1%), and venous thrombotic events (chemotherapy alone: 3.2% versus chemotherapy + bevacizumab: 8.3%). Gastrointestinal and genitourinary bleeding grade 3–4 was uncommon with the addition of bevacizumab (2% versus <1%; p=0.37 and 3% versus <1%; p=0.12). No patient experienced any grade central nervous system bleeding or grade ≥3 congestive heart failure. To date, although the number of cases is small and no definitive conclusions can be drawn, investigation of potential risk factors associated with vaginal fistula or gastrointestinal perforation events in the GOG-240 study identified only prior radiation therapy as a risk factor: 100% of patients who were reported to have gastrointestinal perforation (Standardised MedDRA Queries [SMQ] includes gastrointestinal perforation, gastrointestinal fistula and abscess events) had received prior radiation compared with 80% of the overall population in this study.47
Given the outcome of the disease and impact of treatment-related symptoms, HRQoL is an important aspect to assess in cervical cancer.48 HRQoL was assessed by FACT-Cx, FACT/GOG-Ntx4 and BPI worst pain single item as was previously evaluated in patients with persistent, recurrent or stage IVB carcinoma of the cervix in GOG-204 study. Bevacizumab in combination with chemotherapy did not impact HRQoL as no clinically meaningful changes were observed in the FACT-Cx TOI during treatment and up to 9 months after cycle 1 (99% CI: −4.1–1.7; p=0.30). Patients in both treatment groups reported an increase in neurotoxicity in the FACT/GOG-Ntx4 in the longitudinal analysis. The mean differences observed in neurotoxicity in bevacizumab treated patients were similar to those observed in the chemotherapy alone group.49
Based on the GOG-240 study results, on 14 August 2014, the FDA approved the anti-angiogenesis drug, bevacizumab, for advanced cervical cancer. The approval of bevacizumab for this indication was carried out in record time under the FDA’s new priority review programme.50 Later, on 8 April 2015, the European Medicines Agency (EMA) also approved bevazicumab plus chemotherapy for women with advanced cervical cancer.51
Following the FDA approval, the National Comprehensive Cancer Network (NCCN) upgraded cisplatin-paclitaxel-bevacizumab to category 1 in August 2014 and listed topotecan-paclitaxelbevacizumab as category 1 in September 2014.52 The final analysis from GOG-240 has confirmed that benefits obtained from the addition of bevacizumab are sustained after 348 events and with a median follow-up of 50 months; bevacizumab-containing regimens continue to demonstrate a significant improvement in OS over chemotherapy alone: 16.8 versus 13.3 months (HR=0.765, 95% CI: 0.62, 0.95; p=0.0068).53 The improvement in OS observed with bevacizumab in combination with chemotherapy to greater than 12 months represents a landmark improvement in outcomes for women with this disease. Beyond this, exciting results obtained in a welldesigned clinical trial, with a well-selected population, our efforts should be focused on extending this therapy ‘in the real world’. We must be able to bring this new therapy with their positive results to the developing countries with the highest figures of ‘unselected women’ suffering from cervical cancer.
While the data from GOG-240 resulted in a change to the standard of care not all women benefited and that benefit was relatively short lived. Identification of predictive biomarkers both for response and for toxicity is desirable if we are to optimise the use of this drug. Initial reports from correlative studies evaluated the impact of pretreatment circulating tumour cells (CTCs) on OS showing a correlation between high pretreatment CTC counts, and greater declines of CTC during treatment, with lower risk of death (HR=0.87; 95% CI: 0.79, 0.95) upon addition of bevacizumab.54 Data from this and from other correlative studies are required and validation is essential if predictive biomarkers are to become clinically useful. There are potential opportunities to explore predictive biomarkers across tumour types and data sets, which may benefit a larger number of patients, particularly in relation to prediction of toxicity.
While the addition of bevacizumab to chemotherapy has become a new standard of care for women in resource-rich communities it remains inaccessible to those at greatest need. The cost implications and generalisability of incorporating bevacizumab in poorly resourced countries and communities is a significant
concern; however, questions around cost-effectiveness have also arisen even in resource-rich regions. An initial cost-effectiveness analysis reported by Phippen et al. showed an incremental costeffectiveness ratio (ICER) of $155,000 (€142,000)/quality adjusted life year (QALY);55 however, the static nature of this model results in an oversimplification. Later on, a more sophisticated analysis was developed using a Markov decision tree model to perform a cost-effectiveness analysis of chemotherapy versus chemotherapy plus bevacizumab for treatment of recurrent/persistent or metastatic cervical cancer incorporating the final OS and toxicity data (improvement of 3.9 months and fistula rate of 8.2%) from the GOG-240 study. This analysis showed that the estimated total cost of therapy with bevacizumab is approximately 13.2 times that for chemotherapy alone. In terms of the OS advantage described by the Markov model, an average gain of 3.5 life months will cost an extra $73,791 (€67,580) – an incremental ICER of $21,083 (€19,303) per month of added life – mostly due to the cost of bevacizumab rather than related with the management of related toxicity.56 Before considering if the benefit added by bevacizumab is worthwhile or not from a cost-effectiveness perspective, we should take into account the clinically meaningful results of adding bevacizumab and the singularity of cervical cancer population that most of the time are not able to receive further therapies. Therefore, whether the benefit conferred by bevacizumab is worthwhile or not remains a societal and clinical dilemma. Further exploratory analysis also suggested that adding bevacizumab would become cost-effective, with a significant decline in the ICER, by either reducing the dose of bevacizumab from 15 to 7.5 mg/kg, or diminishing the costs of bevacizumab. In addition, since the current cost-effectiveness model results from a singular data set, the GOG-240 study, namely, the only randomised controlled clinical trial with bevacizumab in cervical cancer, further cost-effective analyses based on real world experience, are warranted. In addition the introduction of generic drugs into the market may in time reduce the cost of targeting angiogenesis using this approach. However, this will not remove the need to advocate on a global level for accessible healthcare for our most economically vulnerable patient populations.
Summary and future directions
Angiogenesis is central to cervical cancer development and progression. The results of GOG-240 study demonstrate a statistically significant and clinically meaningful benefit in OS, PFS and ORR when bevacizumab, an anti-angiogenic agent, is added to chemotherapy in the treatment of
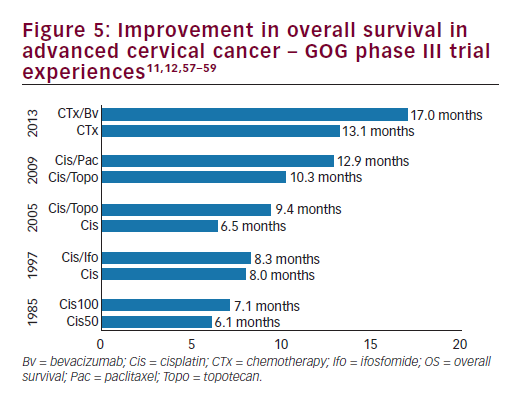
women with persistent, recurrent or metastatic carcinoma of the cervix. The improvement in OS observed with bevacizumab in combination with chemotherapy to greater than 12 months represents a landmark improvement in outcomes for women with this disease and even is most clear when placed in context with prior clinical trials in this setting (see Figure 510,11,57–59 and Table 3).10,11,12,14,57 The HRQoL data demonstrated that the addition of bevacizumab to chemotherapy did not meaningfully impact patients’ QoL in this setting. Therefore, GOG-240 has confirmed that targeting angiogenesis is a successful strategy in cervical cancer that should be further investigated in the next generation of clinical trials and in the different settings of cervical cancer treatment.
Currently, several questions remain open around what is the optimal use of bevacizumab in cervical cancer such as the role of bevacizumab as maintenance therapy and its combination with the carboplatin/ paclitaxel regimen the most discussed. In the GOG-240, per protocol, patients received chemotherapy/bevacizumab until progression, prohibitive toxicity or CR. It was not allowed to stop chemotherapy after a specific number of cycles and continuing bevacizumab alone as in the ovarian cancer trials, namely, OCEANS,60 GOG-21861 and ICON- 7.62 This fact is relevant, given the potential improvement in QoL from discontinuing cytotoxic agents and perhaps without a detrimental in outcomes. We should address bevacizumab as maintenance therapy in prospective clinical trials in order to endorse this approach.
On the other hand, the JGOG-0505 clinical trial14 established noninferiority of the better-tolerated combination of carboplatin and paclitaxel compared with cisplatin-paclitaxel, reinforcing the already extended use of this regimen in the treatment of cervical cancer. Even though the combination of bevacizumab and paclitaxel is safe in other gynaecological cancers, to date this combination has not been addressed in cervical cancer prospectively. In order to cover this unmet need, the CECILIA study (ClinicalTrials.gov Identifier: NCT02467907) has been launched. The trial is a multicentre, single-arm, interventional trial on recurrent, metastatic cervical cancer. One hundred and fifty patients will be treated in 43 sites across Latin America and Europe. This study, recruiting patients since July 2015, aims to assess safety as defined by the frequency and severity of gastrointestinal perforation/fistula, gastrointestinal-vaginal fistula and genitourinary fistula in patients treated with bevacizumab 15 mg/kg in combination with paclitaxel and carboplatin, all repeated every 3 weeks, for recurrent, persistent or metastatic cervical cancer. In addition, this study will include evaluation of the overall safety profile of bevacizumab in combination with paclitaxel and carboplatin in this setting and evaluation of efficacy.
Evidence from the GOG-10934 and a number of other studies showed that improved oxygenation and tumours with higher MVD led to better outcomes with chemoradiotherapy. The combination of an antiangiogenic agent, which promotes vascular normalisation and improved oxygenation combined with multimodality therapy could potentially lead to better outcomes. However, data concerning anti-angiogenic agent/ radiotherapy combinations in other tumour types suggest increased risk of fistula formation, already a concern in women receiving bevacizumab in recurrent and metastatic disease. The Radiation Therapy Oncology Group (RTOG) 1704 evaluated the safety and toxicity profile of adding bevacizumab (10 mg/kg every 2 weeks) for three cycles to pelvic chemoradiotherapy and brachytherapy. In 49 untreated patients with locally advanced cervical cancer (stage IB–IIIB), with a median follow-up of 3.8 years, the 3-year OS was 81.3% (95% CI: 67.2–89.8%) and the 3-year locoregional failure was 23.2%. These outcomes compare favourably with historical reports. In addition, the combination was associated with minimal protocol-defined toxicity, the most common toxicity being myelosuppression. Of note, there were no grade 4 gastrointestinal toxicities or gastrointestinal fistulas or perforations.63
Beyond bevacizumab, exploration of novel anti-angiogenic agents targeting parallel angiogenesis related pathways have been undertaken and considered in women with cervical cancer. Single agent, orally administered, tyrosine-kinase inhibitors (TKIs), pazopanib (VEGFR 1, 2 and 3; PDGF receptor (PDGFR)-α and β; and c-KIT inhibitor) and sunitinib (VEGFR 1, 2 and 3; PDGFR, c-KIT and FLT3 inhibitor) have been investigated. Briefly, some of our first data in anti-angiogenic therapy came from a phase II randomised study that compared pazaponib and lapatinib (a dual TKI of EGFR) to the combination (lapatinib plus pazopanib) published in 2010.64 In this study, 230 patients with stage IVB persistent/recurrent cervical carcinoma not amenable to curative therapy and at least one prior regimen in the metastatic setting, were randomly assigned to one of three arms. Unfortunately, following an interim analysis that showed that the futility boundary for toxicity was crossed, the combination arm was closed. Then, the primary endpoint was PFS for pazopanib versus lapatinib. The study met its primary endpoint and patients treated with pazopanib (n=74) had a slightly longer PFS than those who received lapatinib (n=78) (18 versus 17 weeks, HR=0.66; 90% CI: 0.48–0.91; p=0.013) and OS (HR=0.67; 90% CI: 0.46-0.99; p=0.045) compared with lapatinib alone. Median OS was 50.7 weeks compared with 39.1 weeks for pazopanib and lapatinib, respectively. However, the final OS analysis published in 2011 showed no differences between the two arms. We have to point out that the study was not powered for OS.65 In spite of these data, the development of pazopanib in cervical cancer is no longer active as far as we know. Sunitinib, tested in a phase II clinical trial in patients with unresectable, locally advanced or metastatic cervical carcinoma, was associated with an unacceptably high (26%) rate of fistula formation combined with only modest activity (no documented objective responses and median time to progression of 3.5 months therefore further investigation was not warranted.66
Recently, the CIRCCa trial presented in the 2014 European Society for Medical Oncology (ESMO) Congress67 evaluated cediranib (AZD2171), a selective, orally bioavailable TKI of VEGFR-1, 2 and 3, in 69 women with primary metastatic or relapsed cervical cancer. In the CIRCCa trial, patients were randomised (1:1) to receive three-weekly carboplatin plus paclitaxel, for a maximum of six cycles plus cediranib (20 mg/day) or placebo concurrently with chemotherapy, and later as maintenance therapy until progression. The addition of cediranib improved median PFS by 5 weeks (8.8 versus 7.5 months; p=0.046) and response rate by 24% (p=0.03). However, as CIRCCa closed prematurely owing to the cessation of commercial production of cediranib, the statistical analysis of the difference in median OS between the two groups was underpowered for comparison (59 versus 63 weeks; HR= 0.93; 80% CI: 0.64–1.36; p=0.401). However, the addition of cediranib significantly increased the rate of diarrhoea grades 2–4 (50% compared with 18% in the placebo group (p=0.005) and hypertension (34% versus 12%; p=0.038, respectively). Although the PFS data from the cediranib-containing arm are exciting, the regimen should be modified in order to improve its toxicity profile since the incidence of diarrhoea is greater than expected in a palliative setting. In addition, a future cediranib trial design should take into consideration a bevacizumab-containing regimen as comparator arm.
We are also expecting the results from the nintedanib trial (ClinicalTrials. gov Identifier: NCT02009579), which is a randomised phase II trial comparing thrice-weekly carboplatin (area under the curve [AUC] 5) plus paclitaxel (175 mg/m2) with or without concomitant and maintenance nintedanib (formally, BIBF1120; TKI of VEGFR, fibroblast growth factor receptor [FGFR], PDGFR) in advanced or recurrent cervical cancer. The trial was open in March 2014 and estimated primary completion date is December 2016. Brivanib, another TKI that targets VEGFR2 and FGFR-1, is being evaluated in a phase II study (NCT01267253) conducted by the GOG.
In addition, non-VEGF-dependent therapeutic approaches, including angiopoietin inhibitors, involve other classes of potentially attractive anti-angiogenic drugs and are under investigation in other tumour types. These should also be explored in cervical cancer patients. Furthermore, given that angioprotein-2 (Ang-2) promotes the proangiogenic action of VEGF, the inhibition of Ang-2 and VEGF together could have complementary actions, thus, the combination of an angiopoietin inhibitor, such as trebananib (AMG386), and an agent, such as bevacizumab, could be more active than either agent alone. Combining anti-angiogenic agents with drugs which target the PI3K/ AKT/mTOR pathway may also offer a unique treatment opportunity.
In addition, the significant increase in OS with the addition of bevacizumab to chemotherapy and the subsequent approval by several regulatory agencies will increase the uptake of bevacizumab, creating a new population of advanced cervical cancer patients with a potential window of opportunity through which patients may be considered for participation in clinical trials. Nowadays, the most interesting clinical trials are those investigating the role of immunotherapies and immune system modulators in this patient population. The development of therapeutic vaccines targeting HPV-transformed cells,68 adoptive T-cell therapies69 or immunotherapies involved in immune modulation, including checkpoint inhibitors, is under investigation and represent a powerful novel strategy to improve advanced cervical cancer patients outcomes. Potentially, combining these approaches with an anti-angiogenic agent may also represent a novel non-chemotherapy therapeutic opportunity for this patient population.
As more data emerge about the genomic landscape of cervical cancer and its ‘potentially druggable’ mutations, rational combinations with anti-angiogenic agents will potentially be identified. However, as with all rare cancers, it is vital that any studies undertaken have a strong underlying rationale and that they are designed to maximise the biological information we can learn from them.
Conclusions
Despite the introduction of screening and vaccination programmes, cervical cancer remains a significant health problem. The results from the GOG protocol 240 and the FDA and EMA approval of bevacizumab in combination with chemotherapy for the treatment of women with advanced stage, persistent or recurrent cervical cancer has established the role for new target therapies in a population with historically limited options. However, in order to optimise the use of this agent we need to learn more about patients at risk of toxicity and explore opportunities for developing predictive biomarkers. Moving forward there is a very strong rationale for further exploration of angiogenesis pathways alone and in combination in cervical cancer. However, globally, we need to advocate for affordable and accessible therapeutic options for women affected by this disease.