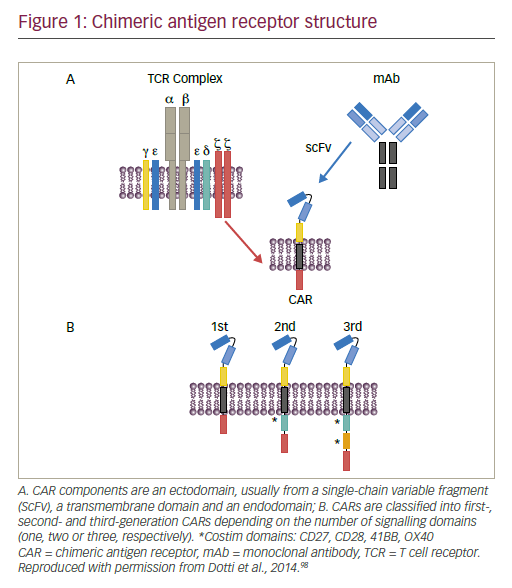
Advances have been made in the use of genetically enhanced T cell therapy, in particular, chimeric antigen receptor (CAR) T cells. Such CAR T cells have been shown to be efficacious in erradicating a number of haematologic malignancies.1–3 CARs are tailored fusion receptors that can combine the specificity of an antigen-specific antibody with numerous downstream signalling domains, the most well-described being T cell activating domains. First developed in the mid-1980s, CARs initially consisted of a variable antigen-binding region of a monoclonal antibody and the constant regions of a T cell receptor (TCR) α and β chains.4,5 Subsequent CARs have been modified to include several functional domains (Rev.6,7, Figure 1) including :
• an ectodomain from a single chain variable fragment (scFv) derived from the antigen binding regions of both heavy and light chains of a monoclonal antibody;
• the hinge region that connects the ectodomain to the transmembrane domain, producing variations in the length and variability of the resulting CAR and affecting its function; a transmembrane domain that is usually derived from CD3-zeta (ζ), CD4, CD8 or CD28 molecules and also has an impact on CAR function;
• an in cis costimulatory domain, a critical component without which the CAR T cell will become anergic upon target encounter. Adding a costimulatory domain activates different signalling pathways and prolongs T cell persistence: two costimulatory domains are included in thirdgeneration CARs to fine-tune the T cell response;
• an endodomain with a signalling domain typically derived from the T cell CD3-ζ chain.
CAR T cells can recognise not only protein, but also carbohydrate and glycolipid structures that are expressed on the tumour cell surface.8 Engineered and endogenous TCRs recognise portions of peptide presented in the context of the major histocompatibility complex (MHC). MHC-independent binding of a CAR to target allows for use of these cells regardless of host MHC polymorphisms. Unlike engineered TCR expressing cells, however, CAR T cells have long been thought to be unable to target intracellular tumour markers. Recently, CARs targeting the extracellular presentation of intracellular tumour markers in the context of a specific MHC have been developed.9,10 This new approach may vastly expand the repertoire of potential CAR targets.
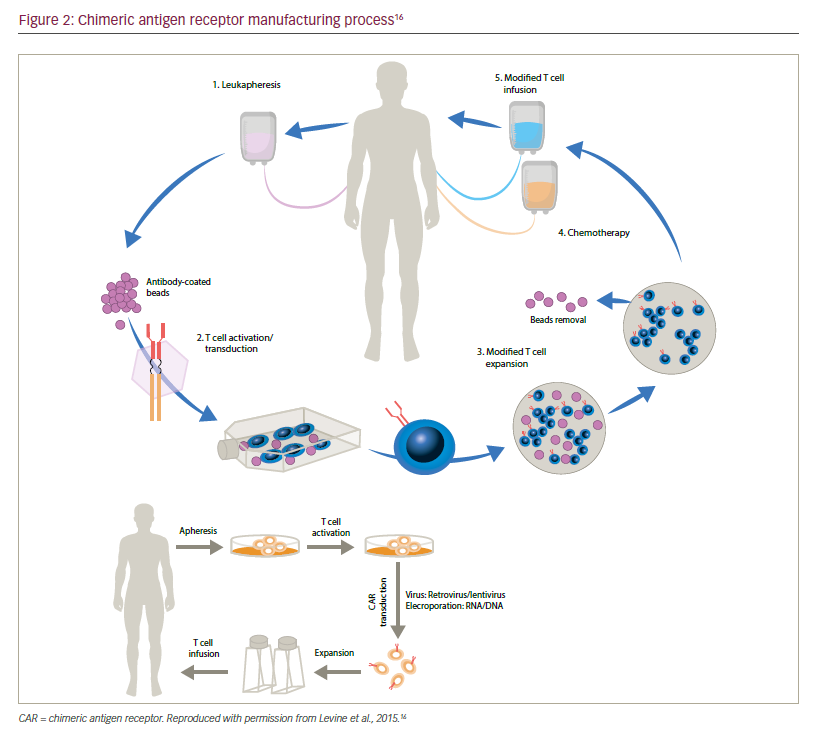
This article aims to describe the process involved in the manufacture of CAR T cells, from the first critical step, harvesting via leukapheresis, to reinfusion, as well as reviewing the clinical evidence in support of the use of CAR T cells.
Generation of CAR T cells
CAR T cell manufacture begins with collection of mononuclear cells (MNCs) from the patient’s blood via leukapheresis (Figure 2). CD3+ T cells may be further enriched ex vivo and then modified to express a transgene encoding a tumour-specific CAR. The CAR-modified T cells undergo ex vivo expansion in optimised T cell culture conditions. The final product is harvested and formulated at a specified dose. Finally, quality and release testing are performed on the product to ensure safety of the infusible product.
Harvesting and isolating T cells for CAR T cell manufacturing
To date, leukapheresis is the most efficient way to obtain significant numbers of T lymphocytes to initiate CAR T cell culture. Apheresis, which is derived from an ancient Greek word meaning “to take away”, describes the process by which whole blood is removed from an individual to be separated into components by centrifugation; subsequently, one or more components are selectively removed and the remainder of the blood is returned to the circulation.11 For the purpose of CAR T cell manufacture, the MNC layer containing lymphocytes and monocytes is the target cell layer to be collected by the apheresis device. Leukapheresis remains the method of choice for T cell collection given that semi-continuous or continuous flow of the blood allows for processing of large blood volumes providing large T cell yields.
It may be challenging to collect MNC products with adequate purity and/ or yield. Monocytes and non-T lymphocytes present in the MNC product can impede downstream CAR T cell manufacturing, and therefore additional purification may be required.12 The MNC layer itself also contains monocytes and non-T lymphocytes including B cells and NK cells in addition to T lymphocytes. Accordingly, MNC products include these cell types to varying degrees. In addition, MNC products may be contaminated with red blood cells, granulocytes and circulating tumour cells or leukemic cells, further complicating downstream manufacturing. Using leukapheresis, it is possible to enrich MNCs from several non-target cells (red blood cells, platelets and granulocytes). In addition, leukapheresis devices such as the Spectra Optia Apheresis® System (Terumo BCT, Lakewood, Colorado, US) offer the ability to further optimise collection technique. Ex vivo, a variety of techniques and devices can be used to further separate or enrich a cellular population from the MNC product. For example, counter-flow centrifugal elutriation (Elutra® Terumo BCT, Lakewood, Colorado, US) is used to separate cells according to size and density based on the cell’s sedimentation velocity. In the setting of excess monocytes, elutriation allows for separating lymphocyte and monocyte cell populations with relatively high purity, good recovery and cell viability.13 Magnetic beads bearing antibodies specific for T cell surface markers can be used to isolate the T cells from the lymphocyte fraction. MNC products with low T cell percentages may benefit from bead-based selection of the target CD3+ T cell population prior to initiation of culture. In addition, beads may be used to activate and expand the T cell population. Finally, traditional density gradients, such as Ficoll, may be used to remove contaminating granulocytes and/or red blood cells. While classically challenging to perform in a good manufacturing practice (GMP)-compliant manner, modern adaptation of Ficoll gradient separation has incorporated closed systems possibly increasing utility in CAR T cell manufacturing.14
Typically, peripheral blood comprises around 20–40% lymphocytes, although circulating lymphocytes can be suppressed in patients being treated for underlying malignancies.15 In addition, most collections occur at steady state, i.e., in the absence of haematopoietic stem cell mobilising agents.16,17 Therefore, processing large blood volumes may be necessary to obtain adequate T cell yield. In this respect, anticoagulant infusion is an important consideration. We opt for apheresis devices where anticoagulant infusion is managed carefully, for instance as with the Optia apheresis device. To pre vent the blood clotting in the apheresis device, an anti-coagulant such as Acid Citrate Dextrose Formula A (ACD-A) is mixed with the blood as it is circulating through the machine.18 As it mixes with the whole blood, citrate binds divalent cations, including calcium, a necessary component in coagulation. The patient is exposed to citrate as the blood components are returned to his/her body and may experience transient hypocalcemia.15 Heparin may also be considered as an anticoagulant; however, this drug is not without its own risk of adverse reaction.19 Several studies have provided strategies to allow larger volume apheresis collections to enhance yield and manage citrate toxicity.20–22
Activating and modifying CAR T cells
Once T cells have been collected, they must be activated, transduced and expanded before being reinfused into the patient. In vivo, endogenous antigen presenting cells (APCs), such as dendritic cells or B cells, may activate cognate T cells. Ex vivo, however, these endogenous APCs display inherent variability making their use impractical in a GMP setting.23 Robust T cell activation for CAR T cell manufacture can be achieved using soluble anti-CD3 monoclonal anitbodies (mAbs),24 anti-CD3/anti-CD28 mAb coated paramagnetic beads25 or cell-based engineered artificial APCs.26,27 Bead-based methods concurrently provide a method to positively select T cells, whereas synthetic cell based artificial APCs allow for customisation of stimulatory conditions. Commercial and technical limitations of bead and APCs respective may make widely available GMP-grade, soluble monoclonal antibody stimulation preferable in some situations. After stimulation, T cells are then genetically modified to express the CAR.
Currently, viral transduction with either gamma retroviral or lentiviral vectors are the most common method of gene transfer owing to the high efficiency of gene delivery and the persistence of integrating vectors in the modified T cells.28 Lentiviral vectors may be preferable
to retroviral vectors due to their ability to transduce non-dividing as well as dividing cells.29 Moreover, lentiviral vectors may have lower oncogenic potential.30,31
Challenges related to viral transduction include the risk of insertional mutagenesis32–34 and ensuring the persistence and expression of the transgene and/or transduced target T cells in the recipient following reinfusion.35 The risk of insertional mutagenesis has been minimised by the use of lentiviral vectors or eliminated by the use of mRNA transfection approaches respectively.29,36,37 While lentiviral vectors are less prone to cause insertional oncogenesis than other retroviral vectors, they nonetheless have the theoretical risk to induce oncogenesis. Specifically, the lentivirus human immunodeficiency virus (HIV) has not been shown to cause T cell lymphomas by insertional oncogenesis, but recent studies show that wild type HIV can insert near oncogenes that clonally expand.38 Nonetheless, it is rational that vectors are less prone to oncogenesis in adoptive T cell therapy compared to stem cell therapy because they infect mature T cells which likely have less oncogenic potential.
The most compelling justification for lentiviral safety in adoptive T cell therapy is the real-world data on HIV infected individuals who are not at increased risk for developing T cell lymphomas. While gamma retroviral vectors appear to have a greater risk of insertional oncogenesis when used to modify stem cells,30–36 the lack of viral induced T leukaemias or lymphomas in patients treated with transduced T cells over a decade ago suggests they can be used safely for T cell therapies.39 A report of longterm data from three clinical studies showed that retrovirus-modified CAR T cells were detectable for more than 11 years following re-infusion. Functionality of these cells has not been confirmed at this time point, however, at the very least persistence of viral transgene expression demonstrates the in vivo durability of construct expression in transduced T cells. Persistence of CAR T cells post reinfusion has been associated with improved outcomes while immune clearance and/or deprivation of proliferative cytokines likely plays a role in loss of efficacy.40,41 Together, these data give hope for long term efficacy with optimised conditions.
Non-viral delivery systems are also being investigated including RNA transfection. RNA transfection leads to transient expression of CAR both in vitro and in vivo.37 While persistence of CAR positive T cells in patients is associated with greater disease control in some cases, long-term persistence may raise concerns for uncontrollable adverse effects. Furthermore, RNA transfection is non-integrating, circumventing concerns over retroviral and lentiviral insertion site oncogenesis.32–35 Finally, repeated doses of RNA transfected CAR T cells may be a viable option,42 though anti-CAR antibodies could limit this approach; a case has been reported of anaphylaxis resulting from CAR-modified T cells, most likely through IgE antibodies specific to the CAR.43 With the development of fully humanised CAR constructs, immunogenicity may be reduced and persistence enhanced.
Expanding modified CAR T cells
Most CAR T cell dosing regimens require a post-modification expansion step to achieve clinically significant numbers of cells for treatment. It is therefore critical to have GMP-compliant methods to expand these cells prior to product harvest. Important factors to consider when expanding CAR T cells ex vivo include (1) provision of adequate nutrient-rich media with or without cytokines throughout the entire culture, (2) optimising gas exchange and (3) operating within a closed system wherever possible. Efficient CAR T cell expansion has been achieved in both static and dynamic culture systems.44 Static culture systems are those in which the culture vessels are stationery and gas exchange occurs via diffusion. Dynamic culture systems add controllable agitation to achieve gas exchange at the gas-fluid interface. In many commercially available static and dynamic culture systems, media exchange can be achieved via heatsealed, closed system addition or removal of media and cytokines. Next generation culture systems may provide real-time monitoring of culture conditions to aid in optimising expansion.
Key challenges within CAR T cell technology
A number of challenges have been identified in the manufacture of CAR T cells. It should be noted that while such issues exist in all aspects of the manufacturing cycle including quality control, product release and logistics, for the purpose of this review, we will primarily focus on challenges in obtaining and growing T cells and in the design of CAR T cells. The manufacture of CAR T cells also frequently proves challenging as autologous cell products obtained from critically ill patients provide unpredictable starting cellular material. In addition, when considering potential CAR T cell targets, one must choose a molecule that allows for maximal tumour clearance while limiting off-tumour effects. Such specific, yet conserved tumour surface markers are limited. New technologies and improvements in existing approaches may allow for next generation CAR T cell products to overcome these challenges.
Overcoming CAR T cell manufacturing challenges
GMP requires that final products consistently meet pre-defined specifications yet, in the setting of autologous CAR T cell therapy, the source material (either whole blood or leukapheresis material) is heterogeneous. There is variability in the numbers of cells harvested from patients due to factors such as disease status and previous treatments; protocols therefore need to optimise the collection, purification and activation of T cell products. Logistics presents another major challenge, with the tracking of patient-specific material through the manufacturing centre. Increasing production capacity while reducing cost is another top priority together with clinical testing.
Current vector transduction protocols involve activating cells to enter the cell cycle and then expanding the cells after transduction in vitro. This process may alter the phenotype of the cells. In the future, it may be possible to transduce T cells with minimal induction of cell cycle by using cytokines such as interleukin (IL)–7 to reduce the relative changes on cellular phenotype. In this manner, T cell subsets such as long lived stem cell memory, naïve, central memory and effector memory will be transduced in their native state.45 However, CAR persistence and expansion in the patient vary greatly between trials.46 Many CAR clinical trials (see discussion in next section) utilised lentiviral vectors that can transduce nondividing cells, which may be advantageous. These vectors potentially transduce nondividing stem cell memory, naïve, central memory and effector memory T cells in their native state as well as CD3/ CD28 activated dividing cells. Differences between CAR signalling motifs also affect persistence in cell culture as well as memory development in the CAR T cell population when reinfused into the patient.47,48 Further, the frequency of T cell memory subsets changes over time.49 In turn, these subsets vary significantly in collected apheresis products, challenging manufacturing standardisation. More work is needed to define the optimal T cell phenotype for CAR T cell manufacturing.
A major difficulty in the broader application of CAR T cell therapy is providing personalised therapy on a large scale. Meeting such challenges will require re-thinking of the CAR T cell paradigm: potential approaches include multi-step CAR (e.g. folate-FITC molecule with FITCspecific CAR T cell described above) or gene-edited cells, engineered to overcome alloreactive barriers through disruption of TCR expression; these are in early stage clinical development. Challenges include collecting sufficient functional T cells from ill individuals who may have a low absolute lymphocyte count and whose existing lymphocytes may be dysfunctional from years of cytotoxic chemotherapy.50 Some patients provide insufficient T cells or T cells that expand poorly ex vivo. Mobilisation strategies that promote collection of lymphocytes might help overcome this challenge. Plerixafor, which is an antagonist of the alpha chemokine receptor CXCR4, when combined with granulocyte colony-stimulating factor results in higher numbers of T cells and T cell progenitors in the collection.51,52 This approach may potentially mobilise CD34+ cells and may affect the activation state or phenotype of the T cells. In addition, there is a need to use manufacturing data to better predict clinical response. Currently, patients who provide T cells that expand well ex vivo may still fail to respond and those with slow ex vivo expansion and minimal cell doses can experience tumour regression as long as expansion and persistence occur in vivo making clinical prediction difficult.53
Target selection to maximise effect and minimise toxicity
Identifying target antigens for CAR therapy may be challenging. Ideally, a tumour antigen should be tumour-specific, i.e., not expressed in healthy cells. However, in many cases, normal and cancer cells are marked by subtle differences in antigen expression. Molecules that are over-expressed in cancer may have roles in cellular transformation and migration. Expression of tumour-specific phosphopeptides or altered glycoproteins may be sufficient to allow tumour-specific targeting.54 Target selection may be particularly challenging for solid tumour target selection because they are typically heterogeneous and most targets are expressed at a low level on benign tissue. Therefore, ideal specific and conserved targets may not exist for a given tumour type. If highly expressed targets are overly prioritised, on-target off-tumour effects are a risk. Similarly, overly prioritising the highly specific targets that are only expressed on a minority of tumour cells poses the risk of selecting target negative tumour outgrowth.
Although early studies of CAR therapy have demonstrated dramatic clinical responses, significant toxicity has also been reported. There are many different types of CAR T cell toxicity, including “on-target, ontumour”, “on-target, off-tumour” and “off-target” toxicity. Neurotoxicity, B cell aplasia, tumour lysis syndrome (TLS), and cytokine release syndrome (CRS) are on-target effects that have been reported in the treatment of patients with CD19-specific CAR T cells. CRS describes a constellation of inflammatory symptoms resulting from cytokine elevations that are associated with T cell engagement and proliferation.55 Symptoms may range from mild and flu-like, to severe, leading to vascular leakage, hypotension, pulmonary oedema, and coagulopathy, resulting in multi-organ system failure. The management of CRS is challenging, since cytokine release is an essential feature of T cell activation and suppressing this may impact on efficacy. One approach targets IL-6, a prominent cytokine in CRS, using the IL-6 receptor antagonist, tocilizumab.55
Recognition of targeted antigens in normal tissues, “on-target, offtumour” toxicity was first reported for the carbonic anhydrase IX (CAIX)- specific scFv expressing T cells.56 CAIX is over- or newly-expressed on tumour cells. CAIX-targeted T cells were used to treat metastatic renal cell cancer but resulted in toxicity most probably caused by the engineered T cells recognising the low level expression of the CAIX antigen in the bile duct epithelial cells. Further study has demonstrated that lowering the dose or shielding benign target positive tissue can prevent such effects.57 In another example of an on-target but offtumour toxicity, a patient with colorectal cancer experienced respiratory distress following a human epidermal growth factor receptor 2 (HER2)- specific CAR T cell infusion and subsequently died from multi-organ failure.58 It was postulated that the T cells recognised HER2 expressed by normal pulmonary endothelium, causing the release of inflammatory cytokines, pulmonary toxicity and a cytokine storm that resulted in multi-organ failure.58 Anaphylaxis has also been documented in patients infused with CAR T cells.59 Off-target effects have been reported with the use of melanoma-associated antigen (MAGE) A3-specific TCR; a peptide from the muscle protein Titin was shown to be an alternative target of the MAGE A3-specific TCR, resulting in fatal toxicity against cardiac tissue.60,61
On and off switches can be employed to potentially terminate in vivo activity of CAR T cells if necessary. One approach involves co-expression of an inducible caspase-9 suicide gene in CAR T cells. After re-infusion, addition of a small molecule can induce T cell apoptosis specifically in the CAR T cells.62,63 Alternatively, control over in vivo function can be achieved through bifunctional small molecules. Such molecules mediate the binding between antigen and CAR, for example, a linker consisting of folate conjugated to fluorescein isothiocyanate (folate-FITC) can redirect and regulate FITC-specific CAR-T cell activity toward folate receptor)-overexpressing tumour cells.64 Finally, CAR T cells have been engineered that have split receptors, in which antigenbinding and all necessary intracellular signalling components assemble only in the presence of a titratable small molecule.65 Regardless of the mechanism, on or off switches add in vivo control over CAR T cell function and potentially enhance safety.
Clinical evidence to support CAR T cell therapy
A growing body of clinical evidence supports the use of CAR T therapy; these are detailed in a number of recent reviews.46,66,67 CD19 is present on B cell leukaemias and lymphomas as well as on healthy B cells, but not on haematopoietic stem cells of other tissues. In one trial, a total of 30 adults and children with acute lymphoblastic leukaemia were given CAR T cell therapy against CD19.1 Of these patients, 27 (90%) achieved complete remission; 20 (67%) achieved a 6-month event-free survival and the overall survival rate was 78%. Durable remissions up to 24 months were observed. In another study, 15 patients with indolent B cell malignancies and diffuse large B cell lymphoma (DLBCL) received a conditioning chemotherapy regimen followed by a single infusion of anti-CD19 CAR T cells.24 A 92% objective response rate was reported in 13 evaluable patients; eight achieved complete remissions, four achieved partial remissions, and one had stable lymphoma. Complete remissions occurred with four of seven evaluable patients with chemotherapy-refractory DLBCL. One patient died due to an unknown cause. In a report on 16 patients with relapsed or refractory B cell acute lymphoblastic leukaemia (B-ALL) treated with autologous T cells expressing the 19–28z CAR specific to the CD19 antigen, the overall complete remission rate was 88% and seven patients successfully underwent allogeneic haematopoeitic stem cell transplantation (HSCT) after CAR T cell therapy.68 This study may lead to a paradigm shift where CAR T cell therapy may occur before attempting allogeneic transplant. In a phase I trial, the feasibility, toxicity, maximum tolerated dose, response rate and biological correlates of response of CD19-specific CAR T cell treatment were defined in children and young adults with refractory B cell malignancies.69 The use of CD19-specific CAR T therapy mediated potent anti-leukemic activity in these individuals. The treatment was deemed feasible and safe: all toxicities were reversible, and prolonged B cell aplasia did not occur. B cell aplasia could potentially be a surrogate marker for CAR-T cell persistence.
Future for CAR T cell therapy
Future directions in CAR-T therapy have the following aims:
• To address current limitations of CAR T cell therapy (efficacy and safety)
• To broaden the effective approaches by seeking new targets
• To develop next generation CARs, with the ultimate aim of producing a universal CAR T cells, i.e., allogeneic “off-the-shelf” products, which may alleviate specific GMP challenges of a personalised therapy.
There is a need for optimisation of lymphocyte collection through improved leukapheresis collection process. Most leukapheresis devices are flexible and adaptable, allowing adjustment of various parameters. A few studies have shown that apheresis parameters can affect the T cell yield and purity.70–72 If it were possible to further enrich lymphocytes over monocytes at the time of collection this would be a welcome advance, as it would minimise the need for further ex vivo manipulation. It is accepted that the number of platelets and red blood cells can be reduced by leukapheresis, but it is less clear whether monocytes and lymphocytes can be separated robustly without further innovation. The Spectra Optia platform now offers two related but functionally distinct MNC collection programs; Optia MNC73,74 and Optia CMNC (Continuous Mononuclear Cell Collection).75,76 Critically, both configurations utilise the automated interface management (AIM) system to identify, address, and continuously monitor and maintain an optimal position for collection of targeted cells located at the interface between packed red blood cells and plasma. During the accumulation phase, the chamber is filling with target cells, while allowing the platelets to continuously move through the chamber and back to the patient. As the chamber becomes full, the smallest cells will exit first (lymphocytes). The operator can control when the chamber flushes during Optia MNC collection. By flushing the chamber before it is full, the operator can influence the lymphocyte content of the product. Variation in lymphocyte content, according to chamber flush, has been reported,71,73 and this variability persists even after the introduction of highly automated apheresis systems.72
Alternative methods of T cell collection for the purposes of CAR T cell manufacturing are under investigation. Nonetheless, to date, leukapheresis remains the standard in the field, primarily due to the procedure’s ability to reproducibly obtain large numbers of viable T cells for initiating culture. Obtaining peripheral blood MNCs via venous blood draw is an attractive method for obtaining MNCs given that it is less invasive than leukapheresis.
However, this method has not yet been proven to be broadly applicable to CAR T cell patients, and specifically lymphopenic patients. While ex vivo expansion is often robust, collection via blood draw compared to leukapheresis will yield fewer T cells and therefore risks not meeting final product dose requirements. In the near future, with improved potency and expansion, these dose requirements may be reduced and allow for starting with fewer cells from a single peripheral blood draw.
Minimal ex vivo manipulation is desirable because each stage is associated with risks for contamination, leading to the use of closed culture systems.77 Furthermore, cell type separation, bead-based purification, and cell washing all decrease yields. Improved bead- or cell-based expansion protocols could reduce the need for ex vivo purification. More advanced T cell modification can also be used to drive CAR T cell expansion ex vivo.78,79 Further, a collection method that greatly reduces platelets, red blood cells and neutrophils could improve the likelihood of obtaining a pure T cell product without heavy manipulation. CMNC may minimise these contaminants more effectively than the standard mononuclear collection protocols.70–72 GMP compliant equipment that utilises closed systems and automation have been developed. However, improved systems to monitor and document the process of generating the CAR T cells are desirable.
In terms of efficacy, inhibitory molecules that interact with activated T cells and their ligands on tumour cells to reduce T cell function include T cell membrane protein-3 and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and/or programmed death-1 (PD-1).80 Blocking these interactions may enhance further the anti-tumour activity of genetically modified T cells. In support of this premise, administration of an anti- PD-1 antibody has been shown to significantly increase the efficacy of CAR T cell therapy in vivo.81 The ability to block this interaction could be engineered into the CAR T cell itself but this may raise the risk of uncontrolled autoimmunity.
The localisation of adequate numbers of CAR T cells to the microenvironment of solid tumours represents a major challenge and work is already in progress to address this issue. Several approaches may optimise the tumour microenvironment for CAR T cell-based immunotherapy, e.g. pre-emptive chemotherapy and use of CAR T cells co-engineered with an inducible cytokine, IL-12. These CAR T cells, when activated by their CAR, deposit IL-12 in the targeted tumour lesion.82 This in turn attracts an innate immune cell response toward those cancer cells that are invisible to CAR T cells. These T cells redirected for universal cytokine-mediated killing (TRUCKs) have shown efficacy against solid tumours with diverse cancer cell phenotypes, promoting interest in their evaluation in clinical trials. However, tumour-infiltrating lymphocytes engineered to express IL-12 have also demonstrated significant clinical toxicities including liver dysfunction, fever and haemodynamic instability associated with high levels of IL-12 and interferon gamma.83 Targeting a minor subset of tumour cells, such as cancer stem cells, may obviate the requirement for extensive T cell recruitment.84,85 Innovative cell delivery systems such as focused ultrasound may also lead to improvements in this area.86
Investigation into a combination of targeted and immune therapies are warranted, for example, a combination of CAR T cell therapy with immunomodulatory protocols to tackle the immunosuppressive microenvironment induced by solid tumours. This approach has shown promise with the demonstration that PD-1/PD-L1 disruption can restore anti-tumour function of CD28 mesothelin-targeted human CAR T cells in orthotopic mouse models of pleural mesothelioma.87 Cherkassky et al. demonstrated that this restoration of effector function could be achieved by PD-1 antibody blockade as well as cell-intrinsic PD-1 small hairpin RNA (shRNA) blockade or PD-1 dominant negative receptor expression, suggesting novel manufacturing pathways to produce CAR T cells immune to PD-1-mediated tumour suppression.87
Sub-optimal T cell trafficking to tumour deposits is another important limitation to the effectiveness of CAR T cell immunotherapy. Future strategies are needed to enhance tumour-specific homing; genetic modifications encoding integrins or ligands may enhance trafficking,88 although most efforts at redirecting T cell migration have focused on chemokine receptors.89 It has been demonstrated that by engineering the CAR T cells to express the chemokine receptor CCR2, enhanced trafficking to the tumour can occur,90,91 suggesting new avenues of research. Traditional chemotherapy upregulates chemokine ligands on tumours, therefore combination regimens may be possible. Furthermore, donor-derived CD19-specific CAR T cells from allogeneic stem cell donors may present an advance in controlling relapse in leukaemic patients and improving the range of applications in adoptive cell therapy protocols.92
Much progress has been made over the past 20 years in the genetic redirection of T cells against cancer and benefits for cancer patients are beginning to be reported. The process of leukapheresis is essential in directing future CAR T cell therapy. Leukapheresis should be tailored specifically to the needs of the CAR T cell manufacturing process and much can be achieved in terms of optimisation. Consistent and/ or optimised collection via leukaphereis is a critical first step of the manufacturing process.
Other future areas of research include: non-CD19 targeted CARs, and transposon systems such as the ‘Sleeping Beauty’ technique that allows the targeting of B cell malignancies.93 In addition, CAR-transduced regulatory T cells (Tregs) are being investigated in non-oncology settings including autoimmune disorders and haemophilia94,95 and CAR T cells that are specific to HIV infected cells.96,97
Summary and concluding remarks
The treatment of cancer patients with CAR T cells is one of the most promising adoptive cellular therapy approaches, and combines the specificity of gene therapy with the wide-ranging response of cellular therapy. However, reproducible manufacturing of high-quality, clinical grade CAR T cell products is necessary for the widespread application of this technology. Leukapheresis is key to the success of CAR T cell manufacture. Further development of CAR designs is needed to achieve more selective killing of malignant tissue and improved activity against cancer target antigens to avoid tumour escape and relapse.