Myelodysplastic syndrome (MDS) is a cytogenetic, epigenetic, and immunological heterogeneous group of myeloid disorders characterized by dysplastic hematopoeisis and propensity for acute myelogenous leukemia (AML) transformation.1,2 The Surveillance, Epidemiology, and End Results (SEER) Program estimates that risk factors for the disease include, among others, aging,3 which is frequently associated with myeloid skewed hematopoeisis,4 male gender, and less characterized, smoking exposure.5 The revised international prognostic score system (R-IPSS) segregates five subgroups with distinct biologic behavior; however, low risk (very low and low) and high risk (intermediate, high, and very high) categories allow risk stratification for therapy selection.6 For patients with low-risk disease, gene expression profiling (GEP) highlights defects leading to immune deregulation, and enhanced apoptosis.7 On the other hand, patients with high risk disease exhibit GEP associated with checkpoint abnormalities and core oncogene activation, such as MYC.8,9
The World Health Organization (WHO) classification of myeloid disorders allocates disease subgroups that integrate MDS with either multilineage dysplasia (MDS-MLD), MDS single lineage dysplasia (MDS-SLD), MDS with single lineage dysplasia and ring sideroblasts (MDS-RSSLD), MDS with multilineage dysplasia and ring sideroblasts (MDS-RSMLD), MDS with excess blasts-1 (MDS-EB1), and MDS with excess blasts-2 (MDS-EB2).10 About 70% of MDS patients would present with anemia, however, quantitative and qualitative defects in granulocytes are commonly observed. Neutrophils play an important role in innate defense mechanisms against bacterial infection. Several previous reports highlight not only an increasing incidence but also significant severity of infection in patients with MDS.11 It is likely that similar defects that initiate dysplastic erythropoiesis originate functional neutrophil abnormalities. In addition, MDS can present with thrombocytopenia, and even functional platelet abnormalities.12,13 Trilineage quantitative defects emphasize the possibility of multiprogenitor stem cell as initiating event. However, full characterization of MDS stem cell/progenitor has been limited by inability to accurately engraft and propagate human disease in xenograft model. This suggests that the bone marrow microenvironment facilitates initiation and maintenance of the disease. To date, MDS remains enigmatic. It is likely that as novel genomic diagnostic techniques develop, further characterization of MDS-initiating events will improve design and delivery of precision therapy. In this article, we will review the current molecular, inflammatory, and infectious basis for dysplastic hematopoietic failure.
Normal hematopoiesis
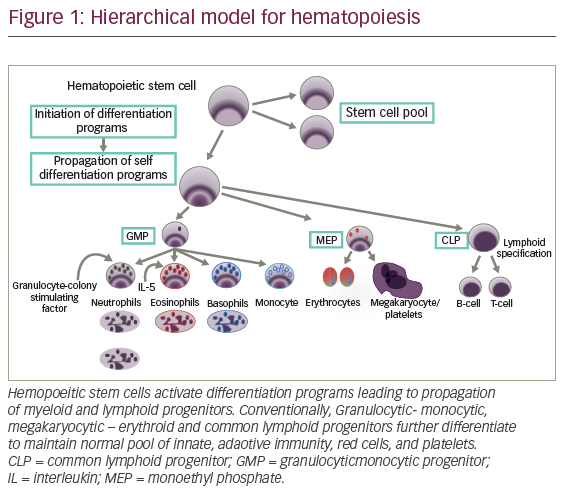
Normal hematopoeisis is a stepwise process that leads to generation of functional red cells, platelets, and granulocytes. Hemopoeitic stem cells (HSCs) proceed through multiple divisions to ensure lifelong provision of differentiated myeloid-lymphoid progenitor (MLP) equipped with potential for stable propagation of self-differentiation programs (see Figure 1). For human blood hierarchy, xenotransplantation has provided significant progress in understanding normal and malignant hematopoiesis.14,15 However, given the small number of HSC (1 x 106 cells), study of stem cell biology has been challenging. These cells reside as dormant fractions (G0) in marrow niches. Initial experiments involved injection of human bone marrow cells into immune-deficient sublethally irradiated mice. Myeloid repopulation was favored by addition of interleukin-3 (IL-3), granulocytic-macrophage-colony stimulating factor (GM-CSF), and stroma cell factor (SCF).16,17 The potential for long-term repopulation (more than 12 weeks) and transient engraftment suggested two subgroups defined as long-term HSC (LT-HSCs) and short-term HCS (ST-HCSs).18 A well-defined C34+ cell is endowed with self-renewal capacity and potential for long-term marrow repopulation.18 HSC division would be expected to result in either cells retaining stem cell program, or changes in GEP consistent with cell differentiation and progressive loss of CD34 expression. Generally speaking, human lineage commitment generates lymphoid subsets including T, B, and natural killer (NK) cells equipped for pre-specified immune response. In addition, fully developed granulocytes such as neutronphils, eosinophils, mast cells, basophils, and monocytes differentiate from granulocytic-monocytic progenitor (GMP) (see Figure 1), whereas erythrocytes and megakaryocytes arise from progenitors with megakaryocyte and erythroid cell potential. Precursors/progenitor terminal differentiation is supported by highly complex interaction between transcription factors, cytokines, and niches structure. Among them, a well-known homeostatic granulocyte-colony-stimulating factor (G-CSF) facilitates granulocytic differentiation. On the other hand, during malignant transformation, myeloid biased progenitor expansion represents a priming event. In addition, adult stem cells/progenitors interface with marrow niche, which also participate in HSC malignant transformation.
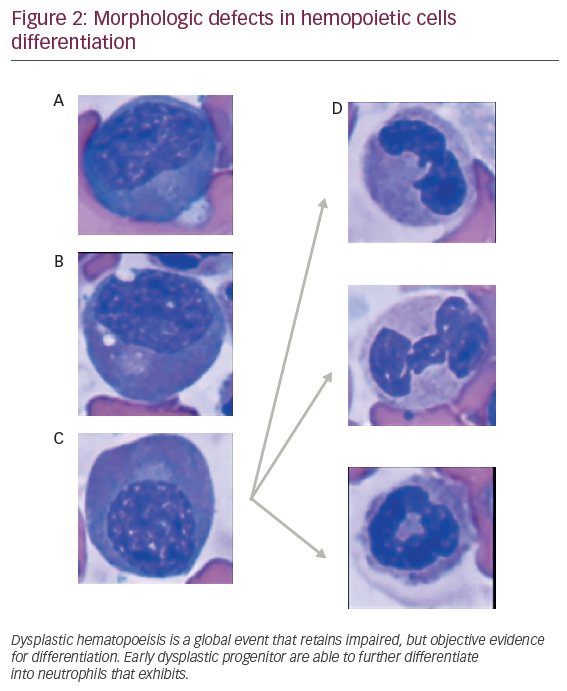
Dysplastic hematopoeisis
The observation that dysplastic myeloid features are retained in different maturation stages in MDS patients suggests a potential hierarchical model for dysplastic hematopoeisis (see Figure 2). Despite difficulties for recapitulating MDS disease in animal xenograft, previous models have demonstrated that coinjection of CD34+ hemopoeitic cells, along with in vitro expanded mesenchymal stromal cells (MSCs) into marrow cavity, faithfully reconstitute human MDS cells. “Disease propagating cells” were restricted to CD34+ CD38- fractions suggesting that disease could occur, at least in a proportion of patients, in multipotent stem cells.19 Supporting this, non-leukemic cell populations, such as lymphocytes obtained from DNMT3Amut AML patients in remission (3 out of 14 patients [21.4%]), harbored similar mutations found in leukemic cells. This mutation were observed at lower allele frequencies (4–31%),20 suggesting a multipotent stem cell disease initiating effect.21 These mutations, originated in the HSC compartment, can clonally expand and progress as preleukemic fractions, contribute to acute AML, an evolving concept that highlights a hierarchical model for MDS.
Aging bone marrow
The physiology of aging hematopoiesis represents an attractive research field given complex interaction between bone marrow microenvironment and HSC. During aging, a gradual functional decline of diverse cellular subsets with different self-renewal and differentiation potential is dominant. However, it is recognized that a shift towards preferential myeloid biased hematopoeisis is a common event that precedes disruption of normal marrow homeostasis.22 Among others, low transforming growth factor (TGF)-β bone marrow concentration provides signaling to induce myeloid biased hematopoiesis.17 In aging HSC, TGF-β regulated genes, such as Nr4a1, Cepba, Jun, and Junb were found to be down-regulated suggesting that TGF-β participates in HSC development.22 Under preferential myeloid hematopoeisis, several other factors contribute to progressive loss of regenerative marrow potential including deregulation in inflammation. Serum levels of circulating proinflammatory cytokines, such as IL-6 and tumor necrosis factor (TNF-α) are typically elevated twofold to fourfold in the elderly, leading to an entity collectively described as inflamm-aging.23 High levels of IL-6 and TNF-α lead to defective mesenchymal stem cell differentiation, impaired osteoblastogenesis, and exacerbates myelopoiesis.23 In addition, aging greatly increases the risk of accumulating harmful mutations in a myeloid skewed context favoring high incidence hematologic malignancies, a term defined as clonal hematopoiesis of indeterminate potential (CHIP).24 Contrasting this, in otherwise healthy old adults even with detectable mutations, such as DNMT3A, phenotypically normal trilineage reconstitution could be sustained for years without excessive risk for hematologic malignancies.25 In this context of “asymptomatic mutational carriers”, patient-associated genetic, epigenetic, and immunologic defects could govern vulnerability for hematologic malignancies.
Cytogenetics abnormalities
Chromosome 5q deletions
Chromosome 5q deletion (del[5q]) is one of the most studied phenotypic expressions of MDS. This distinctive 5q syndrome is characterized by macrocytic anemia, a normal or increased platelet count, erythroid hypoplasia with micromegakarocytes in bone marrow.18–21 Isolated del(5q) can occur in about 10–15% of patients representing the most common isolated cytogenetic abnormality.26 It clusters within the good cytogenetic group and is associated with improved overall survival (OS) and AML-free survival of 57.8 and 202 months, respectively.27 The most commonly reported deletions fall into one of the following three types of common deleted region (CDR): del(5)(q13q31), del(5)(q13q33), and del(5)(q22q35).18–23 The first CDR 5q32-5q33 is associated with the del(5q) syndrome and better prognosis. Within this CDR are SPARC gene and RPS14, a component of the 40S ribosomal subunit whose allelic haploinsuficiency recapitulates phenotypic expression of del(5q) syndrome in vitro.28,29 The second and more proximal CDR located at 5q31 includes candidate genes CDC25c, EGR1, and alpha catenin (CTNNA1), a putative tumor suppressor gene. Deletion of this locus is associated with a complex karyotype, p53 mutation, and more aggressive disease. Outside of the CDRs, several genes including numerous cytokine genes such as APC, NPM1, IL-3, 4, 5, 9, 13, 17β, and GM-CSF are located on 5q.28,30 del(5q) MDS has unique sensitivity to lenalidomide. Mechanistically, lenalidomide inhibits two dual-specificity phosphatases, Cdc25C and PP2A-C, involved in G2/M checkpoint regulation.31 It also induces cellular apoptosis via the p53 pathway by binding to the CRLA4CRBN E3 ubiquitin ligase, altering its substrate affinity to induce the selective degradation of casein kinase 1A1 (CK1α), a known down-regulator of p53, below haploinsufficiency level29,32 and up regulating SPARC.30
Chromosome 7 aberrations
Chromosome 7 aberrations are the second most common distinct chromosomal abnormality in MDS with an overall frequency of 25–30%.31 Entire loss (monosomy 7), partial deletions of the long arm (del[7q]), and translocations have been described.33 Identified CDR includes the band 7q22 and the more telomeric regions 7q31–32 and 7q36. At the genomic level, inactivating mutations in EZH2 play an important pathogenic mechanism in establishing disease phenotype.34 Generally, chromosome 7 aberrations are associated with more severe cytopenias, and an overall poor prognosis. Del(7q) clusters into the intermediate prognostic group, and monosomy 7 into the poor group.31 Some authors supported this prognostication32,33 while others suggested no prognostic difference between del(7q) and monosomy 7.35 At the therapeutic level, small reports of treatment with hypomethylating agents (HMAs), such as azacitidine and decitabine, are encouraging with preferentially good response in monosomy 7 patients,36 but require larger scale validation.
Chromosome 17 abnormalities
Del(17p) has been primarily observed in therapy related AML and MDS. Overall incidence in MDS is about 0.2–3.7%.27,37 Deletion involving the short arm of chromosome 17 is associated with complex karyotype encompassing del(5/5q) and/or del(7/7q).38 Del(17p13.1) contains TP 53, and its associated disease phenotype is characterized by high-risk biology and enhanced propensity for leukemia transformation.35 The presence of chromosome 17 abnormalities as part of complex karyotype conferred an even worse prognosis than that predicted by the R-IPSS with a median OS of 5.4 months.39 Copy number neutral-loss of heterozygosity (CN-LOH) involving 17p was the most common CN-LOH in patients with newly diagnosed MDS and was associated with increased genomic instability and more aggressive disease.40
Chromosome 3 abnormalities
Inv 3 (q21q26) and t(3:3)(q21q26) have been described in de novo or therapy associated MDS and AML. In MDS, these abnormalities represent about 1% of recurrent cytogenetic aberrations.36 Their particular disease phenotype is characterized by MDS-MLD.41 Chromosome 3 abnormalities cluster to the poor risk group. At the genomic level, MDS and AML with 3q26 rearrangement have revealed over expression of the proto-oncogene EVI1 (ecotropic viral integration site-1)42 associated with impaired myeloid differentiation.43 Up regulation of GATA-2 is also identified, and its overexpression promotes aberrant megakaryopoiesis. Wanquet et al.44 reported an overall response rate of 50% in patients with chromosome 3 abnormalities treated with 5-azacitidine (5-aza) and median OS of 10.6 months.
Trisomy 8
Trisomy 8 is the most common chromosomal gain in MDS with an estimated incidence of about 4.2%.27 Phenotypically, patients present with more than one cytopenia, lower blast percentage, and association with hypoplastic bone marrow features.45–47 Severe cytopenias can result from cellular suppression of normal bystander cells mediated by a deregulated cytokine milieu as result of immune directed cytotoxicity.38,48 Pellagatti et al.9 showed that the 10 most significantly deregulated gene pathways in trisomy 8 MDS are associated with the immune response. Trisomy 8 falls into intermediate risk group by Schanz et al. data, with a reported OS of 23.8 months.27 The Spanish MDS group reported a similar survival between trisomy 8 MDS and normal karyotype MDS for blasts count <5% and a worse outcome for blasts greater than or equal to 5%.47 Not surprisingly, immunosuppressive therapy (IST) is backbone treatment for trisomy 8 MDS.49
Chromosome Y
Chromosome Y loss is reported in 2.2% of patients with MDS.22 This incidence increases significantly in males after 70 years of age and with age in both malignant and pre-malignant conditions. In a subset of patients chromosome Y loss is a clonal event; however in others it is clearly an age related event.50 Prognostic wise, -Y MDS, along with del(11q) clusters into the very good prognostic group with a median OS of 60.8 months and low risk of AML transformation.22
Complex karyotype
The overall incidence of MDS with complex karyotype is about 10–18%.27,39 Patients with greater than three cytogenetic abnormalities account for 2.1% and 7%.27 In the study by Schanz et al., OS was 15.6 months with exactly three abnormalities versus 5.7 months for those patients with more than three abnormalities (p≤0.01).27 In patients with complex karyotype, the presence of aberrations that included chromosomes 5 and/or 7 represented a subset with adverse prognosis between all cytogenetic categories.
Monosomal karyotype
Monosomal karyotype (MK) is defined by the presence of at least two autosomal monosomies or a single autosomal monosomy associated with at least one structural abnormality. MK confers inferior outcome in AML. A similar effect was demonstrated for MDS patients presenting with MK. Patnaik et al.40 evaluated 127 patients with MDS with complex karyotype including 106 with MK, observing significantly inferior survival in the subgroup with MK as compared to those with complex karyotype without monosomies (p=0.01; hazard ratio [HR] 1.9). Additionally, a non-statistically significant trend towards inferior leukemia free survival was demonstrated (p=0.09, HR 2.7). The presence or absence of monosomy 7 and/or monosomy 5 did not affect survival among patients with MK. Interestingly, the outcome of AML patients with complex karyotype after allogeneic stem cell transplant was not influenced by harboring MK, a finding that requires confirmation in MDS patients.
Mutational analysis
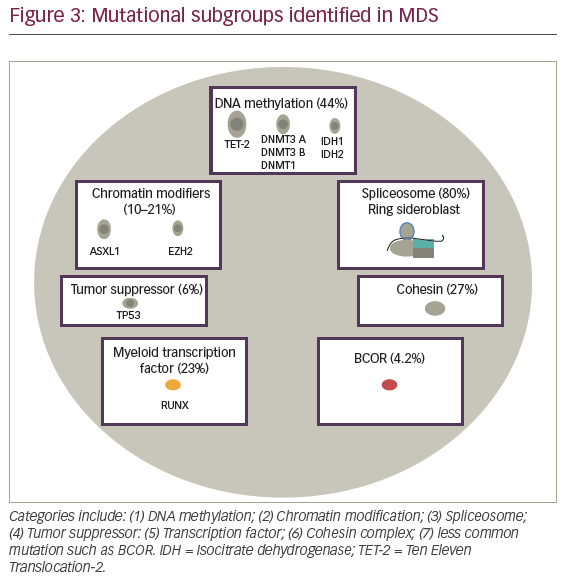
Advances in MDS therapy will derive from molecular precision and identification of potential druggable targets for the disease. In 2005, The Cancer Genome Atlas (TCGA), and during 2008, The International Cancer Genome Consortium (ICGC) were designed as large-scale projects to improve understanding of cancer associated recurrent somatic mutations. In AML, robust prognostic relevant mutations were reported with mutational subgroups frequently observed in MDS suggesting similar ability to detect a significant impact on MDS outcome. Seven subgroups including: activating signaling; DNA methylation; chromatin modifiers; spliceosome, myeloid transcription factors; tumor suppressor genes; and less commonly observed mutations including cohesin complex, BCOR, and BCORL1 (see Figure 3). In this section, we will describe frequency, prognostic implication, and functional implication of frequently observed mutation in myelodysplasia.
DNA methyltransferases-3A (DNMT3A)
DNA methyltransferases (DNMTs) promote conversion of cytosine to 5-methylcytosine (5-mC). Particularly, DNMT3A is active in different biological developmental functions and actively participates in gene expression processes. Enzymatic conditional ablation results in increased self-renewal of HSCs rather than normal differentiation.51 Evolving recent concepts include dominant negative effect in murine embryonic stem (ES) cells, which highlights not only potential for mutated protein to deactivate wild-type enzyme inhibiting methylation, but also reflects a pivotal physiological role in mechanisms of gene expression.52 Somatic DNMT3A mutations have been observed in hematopoietic malignancies with an incidence of 22% in de novo AML.53 OS and event-free survival (EFS) have shown to be inferior in patients harboring the mutation.54 However, in elderly patients with mutated DNMT3A AML, epigenetic treatment was associated with an OS of 15.2 months versus 11.0 months for patients with DNMT3A wild-type.55 As seen in AML, DNMT3A mutation represents an early or founding event in patients diagnosed with MDS. Indeed, mutational frequency of 13% is reported. Previous reports suggest a relationship between DNMT3A defects and an increase in the risk for leukemia progression.56 HMAs are frequently administered in patients with MDS. HMAs are thought to induce inhibition of DNMT1, which improves methylation levels allowing gene re-expression and subsequent differentiation.57 Traina et al. recently described that patients harboring DNMT3A,TET-2, and IDH1 mutations exhibited superior HMA response than similar patients without evidence for the mutations (overall response rate [ORR] for patients with two versus one versus lack of mutations were 75%, 25%, and 20%, respectively).58
Ten Eleven Translocation-2 (TET-2)
The overall incidence for TET-2 mutations in MDS is about 30%.59 Similarly, TET-2 mutations are observed in myeloproliferative neoplasm (MPN) and chronic myelomonocytic leukemia (CMML) in about 10 and 60%, respectively, making this the most frequently mutated gene in hematologic malignancies.60 The biological consequence of TET-2 mutations involves defective conversion of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC). In a normal biologic state TET-2 requires α-ketoglutarate (KG)-Fe(II) and O2 for dioxygenase enzymatic activity in order to mediate 5mC hydroxylation. In addition, TET-2 is involved in chromatin modification through interaction with O-linked b-N-acetylglucosamine transferase (OGT) enhancing the catalytic activity of this enzyme while targeting gene promoters.56 It is possible that TET-2 affects epigenetic modulation of gene expression inhibiting passive demethylation by excluding maintenance DNAMTs.62 Passive demethylation facilitates gene expression in the functional TET-2 state but represses it upon loss of function. TET-2 and IDH1/2 mutations are mutually exclusive. Given α-KG requirement, TET-2 loss of function is normally observed in patients harboring IDH1/2 mutations, in which 2-hydroxyglutarate (2HG) accumulation resulting from isocitrate dehydrogenase (IDH) neomorphic activity, leads to TET-2 inhibition.63 Recently, Traina et al. evaluated 230 patients with MDS, MDS/MPN, or secondary AML (sAML). In the study 92 patients including 53 with MDS, 28 with MDS/MPN, and 11 with sAML were treated with epigenetic therapy. Among them, 17 patients (18%) were TET-2-mutated and showed better azanucleoside response. However, there was no effect from epigenetic therapy on OS and PFS of TET-2-mutated patients.58
Isocitrate dehydrogenase (IDH)1 and IDH2
IDH is a key and rate-limiting NADP+ enzyme for the tricarboxylic acid (TCA) cycle that catalyzes the oxidative decarboxylation of isocitrate to α-KG (2-oxoglutarate). IDH1 and IDH2 mutations are observed in MDS with an incidence of about 5%, and is also observed in other hematologic malignancies like AML in about 15–30% of cases.64 IDH mutations lead to substitution of α-keto by hydroxyl group, in α-KG, converting this metabolite to 2HG, an oncometabolite that competes with α-KG. 2HG interferes with the function of all dioxygenases, such as TET-2.45 IDH mutations can also induce downregulation of the ATM gene by inducing histone methylation,46 resulting in impaired non-homologous end joining repair and thus increasing DNA damage. Both IDH1 and IDH2 have similar incidence in MDS and are thought to be disease initiating mutations. They participate in disease development rather than progression. The prognostic impact for IDH1 mutations has been recently evaluated in a study of 1,782 patients diagnosed with MDS demonstrating that mIDH1 was associated with inferior survival.47 Other smaller studies have demonstrated that mIDH2 decreases overall survival in patients diagnosed with MDS (median, 18.5 versus 31.3 months, p=0.039).58 A growing interest has developed for inhibition of mutated enzyme given leukemogenic transforming potential for 2HG accumulation. The IDH2 inhibitor, AG-221, is one of the current agents used to treat mIDH AML and MDS patients. In a phase I trial for treatment of relapsed/refractory IDH2-mutated AML and MDS patients, the drug has shown promising ORR of 62.5% (complete response [CR] 37.5%). Other agents in development include IDH1 inhibitor AG-120, IDH2-R140Q inhibitor AGI-6780, and IDH1 inhibitor HMS-101.49 In addition, AG-881 represents recent pharmacologic design for dual IDH1 and IDH2 inhibition. This agent is being evaluated in phase I trial and retains blood brain barrier penetration, making the compound attractive for IDH mutated AML complicated with central nervous system (CNS) involvement.49
Enhancer of zeste homolog 2 (EZH2)
Enhancer of zeste homolog 2 (EZH2) is part of the polycomb-group (PcG) and catalytic component of polycomb repressive complex 2 (PRC2), which participates in the regulation of gene expression through DNA methylation and transcriptional repression. This regulatory function of the EZH2 is carried out by introducing a methyl group from the cofactor S-adenosyl-L-methionine (SAM) into histone 3 (H3K27) inducing histone trimethylation.50,66 EZH2 has an important role embryologic development driving X-inactivation, differentiation of lymphocytic cells, and maintaining normal hematopoiesis. Gene mutations resulting in EZH2 overexpression are observed in several cancers, such as prostate and breast. Contrary to this phenomenon, underexpression of EZH2 is a common finding in hematologic malignancies such as MDS playing an important role in disease pathogenesis, especially in patients with deletions of chromosome 7.67 The mutation is normally associated with significantly inferior OS (62 versus 36 months for wild-type versus mutated patients, p=0.033). Overall, mutational frequency is about 10% and is commonly observed in patients exhibiting complex karyotype.67,68 In addition to poor impact on high-risk disease, Bejar et al. found that EZH2 mutations could also be observed in patients with low-risk disease biology resulting in adverse clinical impact. Targeted therapy directed to inhibit EZH2 overexpression and resultant H3K27 hypermethylation has shown to be a potential treatment option not only in hematologic malignancies but also in gliomas and breast cancer. A specific EZH2 inhibitor, GSK-126, has been shown to be active in EZH2-mutated lymphomas, for which activating EZH2 mutations are described.69 However, for this compound, lack of effect is expected in EZH2-mutated MDS/AML, for which mutations are loss of enzymatic function.
ASXL1
ASXL1 is a member of the PcG of proteins that maintains close interaction calypso ortholog BAP1, a deubiquitinating enzyme associated with histone modification and gene repression. However, the specific mechanisms by which ASXL1 mutations participate in hematologic malignancies initiation are not fully understood. Conditional ES cell gene deletion leads to multilineage cytopenias and dysplasia, normally seen in patients with MDS.70 Most of the mutations are heterozygous,71 retaining potential for induction of significant myeloid dysplastic changes, such as pseudo-Pelger huet abnormalities, hypo and hypersegmented neutrophils, and increased evidence of hypogranulated and apoptotic neutrophils.72 Previously, studies have demonstrated that ASXL1 mutation occurs in about 21% of patients diagnosed with MDS.71 Mutations were primarily found in patients with normal karyotype and del(7q/monosomy 7).73 In addition, ASXL1 gene plays an essential role in granulomonocytic development and higher mutation frequencies are observed in CMML than in MDS. In CMML, ASXL1 mutation independently conferred an inferior OS (HR 2.29, p≤0.001).70 The prognostic interaction between ASXL1 and TET-2 was studied by Patnaik et al, demonstrating that the combination of ASXL1 mutation and wild-type TET-2 gene resulted in inferior survival.74 On the other hand, patients with ASXL1 wild-type/TET-mut exhibited superior survival. In a separate analysis, ASXL1 mutation conferred inferior OS when compared to those patients with wild-type ASXL1 (HR 1.74; 95% confidence interval [CI], 1.08–2.82; p=0.024). Importantly, this association was usually restricted to nonsense and frameshift mutations and led to shorter progression time to AML.75
Runt-related transcription factor 1 (RUNX1)
Runt-related transcription factor 1 (RUNX1) also known as AML 1 protein (AML1) is a regulator that plays an important role in the differentiation of lower hematopoietic hierarchies into mature blood cells. The gene is located on chromosome 21 and has a Runt domain, which is a protein motif responsible for DNA binding as a monomer and heterodimerization with core-binding factor β to enhance DNA binding capacity by 10-fold.76 At the genomic level, RUNX1 regulates growth factor genes such as GM-CSF, myeloperoxidase (MPO), and IL-3. Mutations in the RUNX1 gene are highly associated with poor differentiated AML and MDS with a frequency of (23.6%). Point mutations in the RUNX1 gene can be acquired from exposure to radiation or drug-related insults and have a frequency of (42%) in cases of therapy-related MDS and AML, although they have not been associated with an impact on prognosis or therapy sensitivity.73 Chromosomal translocations are also a potential cause of RUNX1 disruption involving transposition with the ETO gene located on chromosome 8q22. t(8;21) translocation generates AML/ETO fusion protein responsible for blast cell self-renewal and their massive accumulation in bone marrow.73 Along with TP53, EZH2, ETV6, and ASXL1, RUNX1 predicted poor outcome in patients with MDS, independently from prognostic risk factors. Another important characteristic of a mutated RUNX1 gene is the role in the progression of MDS to sAML, especially when present at early MDS stage and cooperating events such as FLT3-ITD or MLL-PTD.77
TP53
TP53 gene is located on the short arm of chromosome 17 and encodes for p53, a tumor suppressor protein that activates upon DNA damage. This protein also known as tumor suppresor p53 binds directly to DNA and regulates DNA repair, apoptosis, and cell cycle progression at G1/S regulation point. TP53 interacts with other regulators and genes such as BRCA1 and BRCA2, cyclin dependent protein complexes, and KRAS, among others, making this gene an important target for hematologic and solid malignancies. TP53 mutations are present in 50% of malignancies, and both loss and gain of function have been related to tumorigenesis.78 Mutations of TP53 are seen in multiple hematologic malignancies with a frequency in acute lymphocytic leukemia (ALL), AML, chronic lymphocytic leukemia (CLL), and MDS of 16%, 12%, 7%, and 6%, respectively.78 TP53 mutations are observed in MDS patients harboring 5q deletion and complex karyotype, which is associated with inferior OS and increased risk of AML conversion.79 The prognostic implication for the mutation has been widely reported,79,80 with studies revealing a 5 years cumulative incidence of leukemia development of 77% versus 24%, for mutated versus unmutated TP53.79 Although TP53 mutations are not as frequent in MDS as in other hematologic malignancies, studies have shown that the mutation can be a potential predictor of response to therapy. This was seen in a trial of decitabine reported by Welch et al., demonstrating that in 116 patients, 53 patients (46%) had bone marrow blast clearance (<5% blasts) and response rates were higher among those with unfavorable-risk cytogenetic profile than those with an intermediate-risk or favorable-risk cytogenetic (29 of 43 patients [67%] versus 24 of 71 patients [34%], p<0.001).81 Interestingly, among patients with TP53 mutations, higher responses were observed when compared wild-type TP53 (21 of 21 [100%] versus 32 of 78 [41%], p<0.001).80 Similar findings are also reported by Chang et al.82
Spliceosome mutations
Splicing is a strictly regulated event carried out by spliceosomes that generate protein diversity in cells by removing introns for pre-mRNA to create multiple mature m-RNA from one gene.83 Half of the bone marrow samples from patients with MDS harbor spliceosome mutations. These are considered to be founding mutations that give a selective growth advantage to HSC. Most frequent mutations occur in SF3B1, U2AF1, SRSF2, and ZRSR2. Importantly, all mutations are mutually exclusive with each other, suggesting that the same cell cannot harbor two spliceosome mutations at once.84 Among them, SF3B1 is the first gene associated with specific morphological features of MDS, such as refractory anemia ring sideroblast (RARS).85,86 SF3B1 mutant disease presents with high platelet count, MDS-RSSLD with ≥15% RS in 82.6% patients.85 The phenotypic expression for the disease is normally associated with favorable outcome. However, iron deposition resulting from SF3B1 mutations retains coarse distribution compared to wild-type RARS. At the molecular level, investigation of SF3B1 mutation has provided understanding associated with disruption of splicing factors on stem cell biology. Point mutations result in cell cycle arrest and poor HSC differentiation. Similarly, SRSF2 is a serine-arginine rich splicing factor that has also been recently found to be frequently mutated in 10–15% and 25–30% of MDS and CMML patients, respectively. This mutation is normally associated with poor survival.87 The mutation’s pathogenic mechanism in MDS is not yet fully understood. However, some studies suggest that SRSF2 mutation can lead to increased apoptosis of HSC, alternate splicing of genes, such as CDC25c, and genomic instability.88 U2AF1 is also a serine-rich small auxiliary factor in the splicing machinery of pre-mRNA that interacts with serine-arginine splice factors such as SRSF2. As a spliceosome mutation, it typically occurs in founding clones and has a frequency of 11% in MDS. In the case of U2AF1, the most common mutation is seen on serine codon 34 (Ser34), and it’s not only associated with de novo MDS but also higher risk for sAML.89 Many splicing inhibitors or modulators have been identified. However, it was not until 2016 when the US Food and Drug Administration (FDA) approved H3B-8800, an oral available SF3B1 modulator, for the first human study of spliceosome inhibitors in MDS. This agent is currently in clinical evaluation for MDS, AML, and CMML and has demonstrated to have a selective effect on mutant myeloid cells. 90
Cohesin complex mutations
Cohesin is a protein complex that has a role in many important processes such as chromosome segregation assisting proper chromatid cohesion, organization of genomic architecture, transcription, and gene expression regulation.91 Although the role for cohesin complex in the pathogenesis of hematologic malignancies is not fully understood, this protein is known to be essential for cell division, creating a ring-like structure around sister chromatids, so that proper mitosis can occur.92 The genes that fit in this specific complex are STAG1, STAG2, SMC1A, SMC3, and RAD21, with STAG1 being the most frequently mutated gene. Mutations have been connected with multiple myeloid malignancies, including AML, MDS, and MPNs, with somatic mutations found in 12% of patients and decreased expression in 15%.92 Furthermore, when analyzing the relationship between cohesin mutations and MDS specifically, a cohort study including 1,060 patients MDS (n=386), MPNs (n=55), MDS/MPNs (n=169), and AML (n=450) identified cohesin defects in 11% of low-risk MDS and 17% in high risk MDS patients. Interestingly, STAG2 mutations associated with higher-risk disease (p=0.0023).93 In addition, cohesin defects have been found to represent early events in the pathogenesis of myeloid malignancies, and demonstrate a mutually exclusive phenomenon suggesting that dysregulation of one gene may impact the proper functioning of the entire complex. Cohesin complex defects have been found to have no impact on OS, relapse-free survival (RFS), and CR in AML as proposed by Thol et al.94 However, these mutations were found to be associated with poor OS (27.2 versus 40 months; p=0.023), especially for STAG2 mutant MDS patients surviving >12 months (median survival 35 versus 50 months; p=0.017).93 Some therapeutic approaches have been studied involving therapy with lenalidomide and HMAs for at least four cycles demonstrating that patients harboring STAG2/RAD21 were more likely to respond to HMAs compared with wild-type cases (79% versus 47%; p=0.04). No significant difference in response rate were observed in lenalidomide group (cohesin mutant [50%] versus cohesin wild type [52%] patients).93
BCOR mutations
The BCOR gene codes for a protein that functions as a BCL-6 corepressor associated with an important role in early mesenchymal stem cell development and hematopoiesis. Mutations in the gene have been described in oculofacico-cardiodental syndrome. It has also been identified as novel fusion partner of retinoid acid receptor α in acute promyelocytic leukemia translocation variants.95 Protein function is mediated by DNA-binding at promoter regions of proteins such as BCL-6, more specifically at the poxvirus and zinc finger (POZ) domain, and in part by histone deacetylase activity. BCOR mutations are clonal and disruptive events that result in low expression or loss of function of the protein and have increased frequency in oculo-facio-cardio-dental X-linked syndrome, cytogenetic normal (CN)-AML, sAML, and MDS in less proportion.96 The total frequency of BCOR mutations and other associated proteins such as BCORL-1 in MDS is 4.2%. BCOR defects have been associated with RUNX1 (p=0.002) and DNMT3A mutations (p=0.015). The mutation results in poor MDS prognosis (OS; p=0.013; cumulative incidence of AML transformation: p=0.005).94 More specifically, the deleterious effect of the mutation has been demonstrate after multivariate analysis including factors such as age, IPSS, transfusion dependency, and mutational status (HR 3.3; 95% CI, 1.4–8.1; p=0.008).95
SETBP1 mutations
SETBP1 is a nuclear protein originally discovered to bind to SET and is activated to facilitate granulopoeisis recovery in chronic granulomatous disease.97 SETBP1 mutation has been described in MDS and MDS/MPN overlap with a frequency of 3.8% and 9.4%, respectively.98 This mutation is also associated with sAML (17%) and CMML (15%).97 The gene encoding for the SET binding protein 1 is located on chromosome 18 and is widely distributed in somatic cells.98 Although it has been described that overexpression of SETBP1 impairs the activity of many tumor suppresor genes such as PP2A, the function of SETBP1 protein is not fully characterized. SETB1 mutation are observed in 8.9% of CMML patients and results in an additive independent prognostic value to CMML prognostic score system (CPSS) cytogenetic risk groups.99 SETBP1 mutations are strongly associated with ASXL1 (p<0.001), which has been postulated to enhance risk for AML transformation in ASXL1-mutated MDS patients. All these features suggest that a SETBP1 mutation have a substantial role in MDS pathophysiology and represents a novel mechanism for disease progression, especially among patients presenting with higher white blood cell (WBC) and lower platelet counts and hemoglobin levels.98
Immunity in dysplastic bone marrow
The identification of low risk MDS subgroups with favorable responses to IST led to seminal discovery of deregulated immunity in MDS pathogenesis. This concept was formally emphasized by the recognition that autologous T cells directed cytotoxic effect against hemopoeitic stem precursors in MDS.100,101 Sloand et al. elegantly demonstrated that CD8+ T cells expanded in MDS patients, particularly exhibiting cytotoxicity against trisomy 8 cells, a phenomenon that supported the potential for immunogenicity against MDS precursors.100 With inflammation maintaining a central role in disease initiation, it has become important to decipher the interaction of osteo-hematopoietic niches with innate and adaptive immunity. The mechanisms that govern a fine-tuned tolerance among specialized cellular marrow compartment are unclear. Whether deregulated immunity is the result of eliciting adaptive immune responses against “tumor antigens”, disruption of self-tolerance or a combination of both, remains a clinically unmet research necessity. Interestingly, deregulated adaptive immunity has been reported in low-risk myelodysplasia. In these patients, polyclonal CD4+ T cell expansion associated with high CD8+ T cells oligoclonal/monoclonal frequencies, and markedly, depletion of regulatory T cells (CD4+, CD25+, FOXP3+), results in cytotoxicity against progenitor cells and ineffective hematopoiesis.102 On the other hand, low-risk myelodysplasia retains low apoptotic threshold, prompting high turnover/recycling of endogenous proteins, release of damage associated molecular patterns (DAMPs), and nucleic acids,103 which could serve as ligand for germline encoded innate immunity receptors such as TLR, RIGs,104 and NLRP3 inflammasome. In this section, we describe commonly deregulated cytokines and evolving paradigms that link innate immunity with MDS pathogenesis. Additionally, we summarize current biologic insight for implementation of immune-checkpoint inhibition in MDS.
Abnormal cytokines
Tumor necrosis factor-α (TNF-α)
Abnormal TNF-α level is associated with MDS pathogenesis and disease progression. Previous studies have demonstrated that high level of TNF-α is found in bone marrow; however, it is uncertain whether increased levels originate from macrophages, attracted lymphocytes, stromal cell, or neoplastic clones. MDS is characterized by complex bone marrow inflammatory microenvironment exhibiting abnormally elevated TNF-α level. This has been associated with elevation of vascular endothelial growth factor (VEGF) levels, and increased vascularity/dense microvessel formation characteristic of more advance stages of the disease.105 Gene expression has revealed that leukemia initiating cells (LIC), and probably high risk MDS precursor/stem cells, retain high dependence on TNF-α for NF-kB activation.106
Interleukin-6 (IL-6)
This cytokine has emerged as strong candidate in MDS pathogenesis given its proinflammatory properties.107 IL-6 upregulation is associated with increased proliferation and apoptosis resistance of T-cells, generation of TH 17 secreting T cell fractions, and inhibition of regulatory T cells.108 Immunologic deregulation of this sort suggests a pivotal role for IL-6 in initiation and maintenance of inflammatory signaling in bone marrow. Members of the IL-6-family cytokine are composed of two receptor chains and downstream signaling molecules, such as the common signal transducer gp130, which triggers activation of jaunts-activated kinase (JAK)-STAT3 and JAK-SH2, domain-containing protein kinase (MPAK) pathways that executes activation of the genes encoding suppressor of cytokine signaling-1 (SOCS1) and SOCS3.109 In physiologic conditions, it is expected that SOCS1 and SOCS3 expression would induce normal repressive feedback loop that would terminate IL-6 signaling,110 a phenomenon deregulated in hematologic malignancies.
Interferon-γ (IFN-γ)
Bone marrow cytokine homeostasis contributes to regulate normal development of bone marrow stem cells. However, under elevated interferon-γ (IFN-γ) concentration and chronic inflammation, a profound impact on HSC that recapitulates suppression of proliferation and impaired hematopoietic output can be observed. IFN-γ can upregulate CD95 expression in differentiating erythroid progenitors resulting in sequestration of cell growth and differentiation.111,112 In addition, IFN-γ has a profound effect on increased apoptosis by upregulation and activation of caspases 1, 3, and 8, highlighting the need for targetable immunomodulatory interventions that minimize INF-γ mediated infective hematopoeisis.111
Role of Toll-like receptor
Inflammation represents a common pathogenic mechanism for chronic conditions such as myelodysplasia, rheumatoid arthritis, and depression. In recent years, high expression of toll-like receptor (TLR) 3, 4, and 7 in patients with major depression,113 and TLR1, TLR2, and TLR6 overexpression in CD34+ cells of patients with MDS,114,115 reinforced the concept that amplification of inflammatory response remains central to disease pathogenesis. In macrophages, TLRs and NLRP3 inflammasome amplify inflammation contributing to manifestation of ineffective dysplastic hematopoiesis. TLR-2 mediates, in low risk MDS, NF-kB activation leading to elevated IL-6 and TNF-α levels,116 a concept that potentially capitalizes the unfavorable origin for pro-apoptotic cytokines in low risk MDS. Several mechanisms are thought to polarize TLRs toward harmful response. Among them, gain of function TLR2 F217S mutation has gained significant interest.114 Wei et al. demonstrated that the mutation was associated with significant NF-kB, p38MAPK, and IRAK1 modifications after TLR2 agonist stimulation. In the same study, the author suggested the possibility that TLR polymorphism could result in enhanced propensity for MDS initiation.114 Additionally, it is known that MDS accumulates mutations that result in DNA damage, subsequently stromal/stem cells apoptosis and cytokine release, a phenomenon substantially amplified by tissue-resident macrophages.117 Intuitively, in MDS, TLR activation mediated by conserved molecular patterns released from tissue damage or cell death, could contribute to disease initiation.
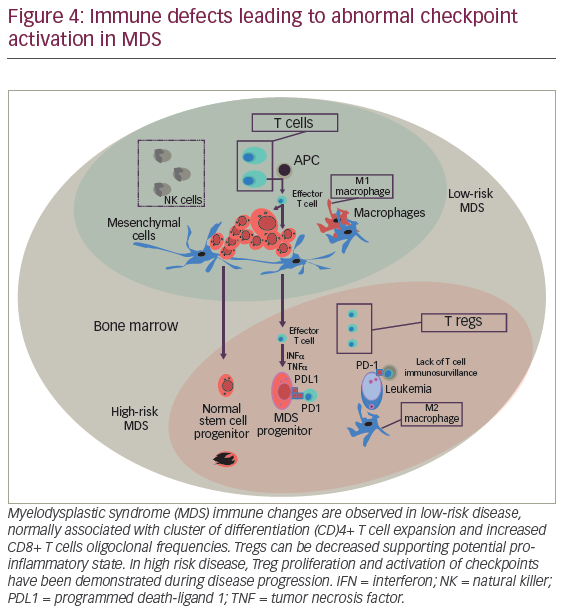
Checkpoint physiology and hematopoiesis in MDS
The mechanisms that direct self-tolerance among specialized hemopoeitic cells are not fully characterized. However, evolving data supports the role of immunity leading to adaptive immune responses against “tumor antigens”. The suggestion that “effective cellular immunity is feasible against hemopoeitic malignancies” is derived from previous observations demonstrating oligoclonal CD8+ T cell expansion, particularly T cells against MDS progenitors harboring trisomy 8. In addition, azanucleoside therapy has been reported to induce autologous immune responses against neo-antigens, such as cancer testis antigens (CTAs) including NY-ESO-1 and the MAGE-A gene family representing a promising intervention to improve tumor immunity.118,119 However, durability, efficacy, and probably quality of T cell responses may be harnessed by adaptive mechanism of “tumor escape”. Similar to solid tumors, hemopoeitic malignancies subvert T cell immunity by activating coinhibitory pathways including immune checkpoint receptors (i.e., cytotoxic T-lymphocyte–associated antigen 4 [CTLA-4] and program death-1 [PD-1]) (Figure 4).120,121 PD-1 is a member of the B7 receptor family, which is involved fine-tuned regulation of immune response. The interaction of PD-1 with his cognate receptor ligands PD-L1 and PD-L2 signal to down-regulate T cell receptor leading to T cell apoptosis. Interestingly, for patients treated with HMAs, significant demethylation of PD-1 promoter in marrow T cells was demonstrated, suggesting that this phenomenon could represent, at least in part, a mechanism for HMA failure. Pharmacological PD-1 inhibition by fully humanized immunoglobulin G-4 (IgG-4) blocking monoclonal antibody redirects and enhances T cell tumor cytotoxity, a concept that holds significant therapeutic hope for AML and high-risk MDS.
Role of infectious disease
HSCs have the ability to proliferate, differentiate, and migrate in order to meet the needs of the immune systems in response to different pathogens. This is a highly regulated process that prevents bone marrow cytokine overexpression and potential HSC exhaustion.122 For decades, immune-mediated T cell against intracellular infections have been thought to participate, at least in part, in stem cell damage. This concept highlights that alterations in different cell lines depend not only on the type of microorganism involved, but also the effect of proinflammatory cytokines released during infection. Overall, the immune response against pathogens allows clearance of the infection, but chronic activation or deregulation of this defense mechanism may lead to detrimental effects.
In general, the stimuli for HSC proliferation and differentiation are led by specialized pattern-recognition receptors (PRRs), which includes TLRs that bind to pathogen-associated molecular patterns (PAMPs).122 Activation of TLRs initiates a cascade of signaling events that promote the release of cytokines and chemokines, attracting immune cells and initiating an inflammatory response, mediated by myeloid differentiation factor 88 (MYD88) and TIR-domain-containing adapter-inducing IFN-β (TRIF). However, chronic TLR4 activation has shown to produce myelosupression, HSC dysfunction, and injury.123
The response against bacterial infection is known as emergency granulopoeisis, and consists in leukocytosis, neutrophilia, production of immature neutrophils and myelomonocytes.122 One of the proposed mechanism is the expansion of bone marrow lineage-negative SCA1+KIT+ (LSK) cells, and progenitor stem cells, after mice infection with Escherichia coli. This effect is primarily driven by lipopolysaccharide (LPS) exposure, and subsequent TNF and nuclear factor kappa B (NFκB) activation.124 Infection also increases G-CSF levels, which stimulates neutrophils mobilization and production allowing selection of early progenitors to differentiate into cells of myelomonocytic lineage.122 Additionally, the suppressive role of IFN-γ in the activation of HSCs has been documented in mice models of mycobacterium avium infection. However, in tuberculosis, it may induce apoptosis of hematopoietic progenitors, leading to BM failure.124 Zebrafish models have proven that infection-induced granulopoeisis is led by transcription factor C/EBPβ, in contrast to steady-state granulopoeisis, in which C/EBPs and C/EBPα are essential.125
Similar to bacterial infections, viruses like cytomegalovirus (CMV) and human herpesvirus (HHV) suppress hematopoiesis after direct infection of HSCs, while human immunodeficiency virus (HIV) and human T-lymphotropic virus (HTLV) produce hematopoietic malignancies like B cell and Hodgkin’s lymphoma by deregulating microRNAs. Aplastic anemia is induced by apoptosis of hematopoietic cells mediated by IFN-γ and TNFα, which is normally associated with parvovirus B19, Epstein-Barr virus (EBV), CMV, varicella-zoster virus (VZV), HHV-6, HIV, and hepatitis A and C viruses (HAV and HCV).124
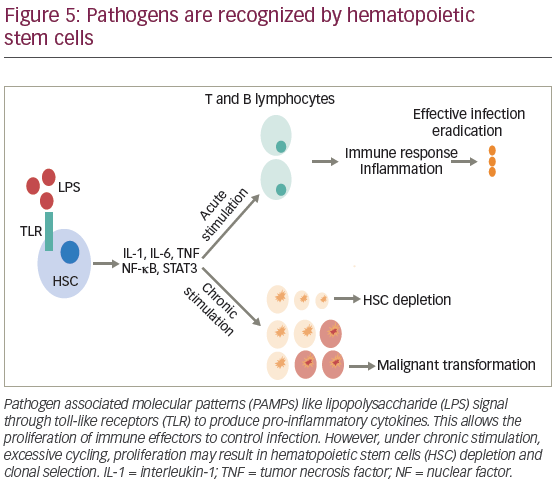
Interestingly, bone marrow microenvironment is also involved in the regulation of HSCs after infection. TLRs are present in nonhematopoietic cells, such as osteoblasts, endothelial cells, endosteal progenitors, reticular cells, and MSC.122 Receptor ligation allows adapting receptor response to infection permitting HSC differentiation during periods of high demand. In the past, infection of stromal cells has been demonstrated to impact hematopoiesis. Specifically, viral infections induced by lymphocytic choriomeningitis virus (LCMV), vaccinia virus, and vesicular stomatitis virus have been associated with high IL-6 expression by non-hemopoeitic cells. Interestingly, upregulation of myelosupressive cytokines has been observed in type B Coxsackie virus infection in mice, which is associated with stroma destruction and decrease in erythroid and lymphoid progenitors mediated by type I IFNs.124
Though HSC activation against infections may be beneficial, there is increasing concern for negative effect given known association between inflammation and cancer.122 Immune cells in the tumor microenvironment, especially macrophages and T cells, have shown to exhibit pro-tumorigenic and anti-tumorigenic functions.126 During acute infection, cytokines like IL-1, IL-6, and TNF stimulates HSC via JAK/STAT inducing NFκB activation.123 In bone marrow, JAK/STAT activation leads to increased angiogenesis and hematopoietic tumor growth (Figure 5).126 Additionally, inflammation also causes local immunosuppression, allowing tumor cells to overcome immunosurvillance by activating immune checkpoint.
Inflammation-induced high HSC proliferation may lead to accumulation of mutations during DNA replication, a phenomenon that can induce genomic instability and epigenetic modifications.122,127 Also, reactive oxygen species (ROS) and reactive nitrogen intermediates (RNI) induce accelerate DNA damage.126 Neutrophil-induced ROS and RNI are important innate immunity effectors that participate in resolution of infections. Similarly, neutrophils engage in tumor progression by increasing secretion of matrix metallopeptidase 9 (MMP9), VEGF, and arginase-1 (ARG1), which inhibits CD8+ T cells.127 Recently, it was identified that neutrophils could favor metastasis by inhibition of T cell and NK cell activity, trapping cancer cells in extracellular traps (NETs), and increasing production of leukotrienes and IL-1β. It is possible that a similar effect prime hematologic malignancy progression. Indeed, case control studies have shown an increased risk for MDS in patients with history of infections. The association is higher with bacterial infections.128 More specifically, TLR-4 has been shown to be overexpressed and constitutively upregulated in the bone marrow of patients with MDS. This receptor is implicated in apoptosis through Fas/Fas ligand pathway, and its expression is regulated by TNFα.122
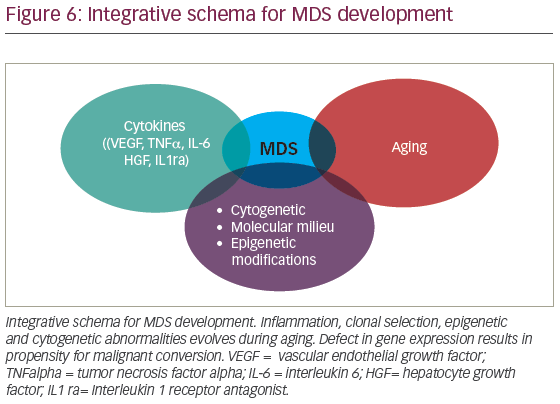
Conclusions and future directions
MDS is conventionally thought to result from a complex interaction of deregulated cytokines, molecular cytogenetic, and epigenetic defects associated with aging (Figure 6). However, in recent years, clonal hematopoiesis of undetermined significance (CHIP) has been recognized as potential initiating MDS event. Detectable myeloid mutations, in the elderly, are associated with significant risk for cardiovascular complications suggesting that profound defects in thrombo-inflammation, development of atherosclerosis, and possibly heart failure are interconnected. Whether chronic inflammation induced by aging (inflammaging) contributes to clonal selection or CHIP exacerbates inflammaging is debatable. With the latter holding potential mechanistic insight, it is then, conceivable that CHIP orchestrates not only inflammaging but broader vulnerability toward malignant transformation. It is likely that we will soon witness deeper understanding of mechanisms that interconnect deregulated innate immunity and clonal hematopoiesis. The role of chronic infections in HSC physiology and clonal selection is an area of significant interest. As we continue to unravel MDS pathogenesis, we will make sense of the increasing disease complexity. In the era of precision medicine, we are expected to better understand how CHIP develops. This will improve design of therapeutic interventions seeking to ameliorate systemic inflammation and overall risk for hematologic malignancies associated with clonal hematopoiesis.