Diagnosis is often delayed in patients with Gaucher disease type 1
Case 1
A 37-year-old Caucasian woman with spherocytosis reported since childhood presented with swollen ankles, gradually increasing abdominal girth, sudden weight gain (3 kg in 6 days), fatigue and exertional dyspnoea. Patient history showed delayed growth during childhood, hepatosplenomegaly from 3 years of age (resulting in an incorrect diagnosis of spherocytosis) and a severe sport-related fracture of the humerus with humeral head necrosis. No alcohol abuse, liver disease/hepatitis, blood coagulation abnormalities or prothrombotic diseases were reported. The first of the patient’s two pregnancies resulted in post-partum bleeding requiring a blood transfusion.
Physical examination revealed pallor, hepatosplenomegaly and oedema in both legs, which resolved with diuretics (a sign of portal hypertension). Blood examination showed leucopenia (1,730/mm3), thrombocytopenia (92,000/mm3) and normochromic, normocytic anaemia (haemoglobin [Hb] 7.8 g/dl, mean corpuscular volume [MCV] 90.1 fl) with mildly increased ferritin (352 ng/ml). Transferrin saturation (TSAT), serum iron levels, haemolytic indices, folate/vitamin B12 and renal and liver function were normal. Autoimmune, virological and neoplastic diseases were excluded. Blood smear and osmotic globular resistance tests excluded spherocytosis.
Splenomegaly (diameter 18 cm) and hepatic stiffness (7.4 KPa; normal value <5 KPa) consistent with stage 1 fibrosis were confirmed. Liver disease (but not Gaucher disease [GD]) was suspected and therefore a liver biopsy was performed. Significant portal and lobular infiltration with aggregated CD68-positive polygonal cells and granulated cytoplasm was observed, with no iron overload. GD was suspected at this point. Further tests showed significantly elevated chitotriosidase activity (14,290 nmol/h/ml; normal value <100), low β-glucosidase activity (28 nmol/h/mg; normal range 200–500) and the identification of β-glucosidase (GBA) gene mutations, all of which led to a diagnosis of GD type 1. Treatment with enzyme replacement therapy (ERT) led to improved laboratory parameters after 1 year. The patient is currently well and continues to receive ERT with no side effects.
The (mis)diagnosis of spherocytosis disguised the patients’ clinical manifestations of GD from childhood to adult life, thereby exposing her to an inappropriate liver biopsy and delaying appropriate management.
Case 2
A 15-month-old child presented with persistent fever, cough and mild respiratory distress. Initial tests showed hepatosplenomegaly, leukocytosis with lymphocytosis (white blood cell count [WBC] 30,130/μl; L 73%), microcytic anaemia (Hb 8 g/dl; MCV 63 fl), thrombocytopenia (114,000/μl) and increased C-reactive protein (CRP; 10.8 mg/dl; normal volunteers <0.5 mg/dl). The child was admitted to the paediatric ward with a suspected infection and was treated with cefotaxime three times daily.
The patient’s medical history revealed growth delays from 6 months of age, previous hospital admissions for fever and a urinary tract infection, normal neurological development (other than a minor speech delay) and no pathological eye movements. Serological and microbiological tests were positive for cytomegalovirus (CMV), but negative for lymphoproliferative disease and Epstein Barr virus. Reduced TSAT (4%) with mildly increased ferritin levels suggested that microcytic anaemia was partially related to iron deficiency. Haemolytic indices were normal.
Fever and respiratory distress gradually improved, CRP levels normalised and CMV infection resolved. However, hepatosplenomegaly persisted (spleen length, 10.9 cm) and anaemia associated with fluctuating, mild thrombocytopenia (113,000–190,000/μl) did not improve after oral iron therapy. A bone marrow aspirate showed no signs of haematological disease; however, a diagnosis of GD could not be excluded based on this finding.
The combination of growth retardation, anaemia, thrombocytopenia and hepatosplenomegaly suggested a lysosomal storage disease (LSD). A dried blood spot (DBS) test showed reduced β-glycosidase activity and molecular analysis detected a homozygous mutation in the GBA gene (c.1226A>G/c.1226A>G; N370S/N370S). GD type 1 was diagnosed and ERT was prescribed. Anaemia and thrombocytopenia resolved after 3 months; hepatosplenomegaly and growth parameters improved up to 1-year post-treatment, with no side effects.
Gaucher disease
GD is a rare, autosomal recessive disorder caused by mutations in the GBA gene (GBA1; EC 3.2.1.45).1–7 More than 330 mutations have been identified to date, all of which result in reduced levels of the enzyme, which in healthy individuals hydrolyses glucosylceramide into ceramide and glucose. GD is characterised by an accumulation of sphingolipids – primarily glucosylceramide and glucosylsphingosine – in macrophages and other reticuloendothelial cells, leading to the formation of ‘Gaucher cells’ (Figure 1). These lipid-laden cells gradually infiltrate the spleen, liver, kidneys, lung, brain and/or bone marrow causing progressive and heterogeneous disease. Although rare, GD is the most common type of LSD, with an estimated prevalence of 1/75,000 births among the general population, rising to 1/600 births among Ashkenazi Jews.5
GD encompasses a continuum of phenotypes ranging from a perinatal lethal form (type 2) to a mild or asymptomatic adult form (type 1).8 The vast majority of patients with GD type 1 present with haematological complications, including spontaneous bruising/bleeding due to thrombocytopenia and/or GD-associated coagulopathy, abdominal discomfort/swelling due to splenomegaly and/or chronic fatigue due to anaemia.2,4,8–10 Other symptoms, including hepatomegaly and abnormal liver function, increased susceptibility to infection due to suboptimal neutrophil function and neutropenia, pulmonary disease, bone pain and (in children) growth retardation and/or delayed puberty may also be present.

GD type 1 (Online Mendelian Inheritance in Man® [OMIM®] number 230800) – the most common and mildest form of the disorder – is usually distinguished from types 2 and 3 (OMIM numbers 230900 and 231000, respectively) by the absence of neurological symptoms. In patients with GD types 2 and 3, these include convulsions, hypertonia, mental retardation, apnoea, slow saccades and/or dementia.4,6,11 However, the distinction between GD types is not always clear. For example, peripheral neuropathies and neurological symptoms resulting from spinal compression fractures have been reported in patients with GD type 1 and a higher incidence of Parkinsonism has recently been associated with mutations in the GBA1 gene.12,13 Conversely, the neuropathic symptoms associated with GD type 3 may be mild, with many patients graduating from college and leading a relatively normal life.14
Long-term complications associated with GD type 1 include pulmonary hypertension,15 haematological malignancies (leukaemia, multiple myeloma and lymphoma), and — less frequently — solid cancers (e.g. hepatocellular carcinoma, melanoma and pancreatic cancer).16–19 Skeletal disorders due to bone marrow infiltration (e.g. pathological fractures, osteolytic lesions and osteonecrosis) are also common and may be associated with a significantly reduced quality of life.2,3,20 Prognosis varies depending on GD type, organ involvement, symptom severity and time from symptom-onset to treatment. In general, patients with GD type 1 who remain untreated survive through to adulthood, whereas those with GD type 2 (<1% of patients) die within the first few years of life, and those with type 3 (<5%) survive into early (and sometimes late) adulthood.4,6,8
The usual treatment for GD type 1 is ERT using macrophage-targeted recombinant β-glucosidase (imiglucerase [Cerezyme®; Sanofi Genzyme, Cambridge, MA, US], velaglucerase alfa [VPRIV; Shire, Lexington, MA, US] and, in some European countries, taliglucerase alfa [Elelyso®; Protalix Biotherapeutics and Pfizer, New York, NY, US).2,10,21 ERT acts by augmenting deficient β-glucosidase activity via intravenous infusions of functional enzyme and has been proven safe and effective for preventing and/or reversing many of the symptoms associated with GD. The magnitude and speed of response to ERT largely depend on the individual patient’s clinical characteristics, symptom severity and rate of disease progression.21 Of particular note is the limited response to ERT in patients with GD-associated malignancies and its lack of effect on neurological symptoms. Another therapeutic approach is to use substrate reduction therapy (SRT) such as miglustat (Zavesca®; Actelion Pharmaceuticals, San Francisco, CA, US) or eliglustat (Cerdelga®; Sanofi Genzyme, Cambridge, MA, US), which can alleviate non-neurological GD symptoms by reducing glucocerebroside production.2 In Europe and the USA, eliglustat is approved as a first-line therapy for adults with GD type 1 who, based on CYP2D6 genotyping, are predicted to be extensive, intermediate, or poor metabolisers. This constitutes >90% of patients with GD type 1.21 Miglustat is indicated for the oral treatment of adult patients with mild to moderate type 1 Gaucher disease and may be used only in the treatment of patients for whom ERT is unsuitable.
The importance of an early diagnosis and prompt management
Prompt treatment of GD type 1 with ERT can prevent or reverse many of the clinical features associated with this condition, thereby improving quality of life.1,3,9,20,22–29 For example, a study of 884 patients with GD type 1 found that, when initiated during childhood, 8 years’ continuous treatment with ERT was associated with the normalisation/near-normalisation of Hb levels, platelet count, liver and spleen volumes, and mean bone mineral density z-score, with significant reductions in the incidence of bone crises.24 Conversely, a study in 17 patients with GD with delayed diagnosis (up to 10 years) found that approximately 25% developed irreversible complications including avascular bone necrosis, severe bleeding, chronic bone pain, life-threatening sepsis, pathologic fractures, growth failure and liver pathology.30 In addition to enabling effective management strategies, a prompt diagnosis of GD enables early screening of at-risk relatives and the timely provision of genetic counselling for affected individuals and their families/carers.
Despite the importance of early symptom management, people with GD often experience considerable diagnostic delays.30–32 Most patients with early GD symptoms are initially referred to a haematologist and/or paediatrician.32 However, a survey of 406 haematologist–oncologists from seven countries found that only 20% of physicians included GD in their differential diagnosis of patients with classic GD symptoms, with most suspecting leukaemia, lymphoma or multiple myeloma.30 Of the 136 patients followed in this survey, the average time from symptom development to diagnosis was 4.1 ± 10.3 years. According to a retrospective review of 86 GD cases from a single centre, 19% of patients experienced diagnostic delays of longer than 5 years.31 Perhaps surprisingly, the Gaucher Outcomes Study (GOS) found that, of the 1,209 participants with diagnosed GD, at least 174 (14.4%) patients remained untreated.9 Whereas some of these patients may have had mild or absent symptoms that (physicians believed) did not warrant expensive pharmacological treatments, others may not have had access to required medications. Nonetheless, results suggest that management delays in patients with GD might be confounded by a lack of awareness regarding the benefits of prompt treatment.
Diagnostic clues
Approximately 60% of patients with GD type 1 have thrombocytopenia at diagnosis and 86% (and 95% of children)8 have splenomegaly.30 Suspicion of GD should be further raised by the presence of hepatomegaly, fatigue (sometimes related to anaemia), leucopenia, bone pain/disease, growth retardation and/or delayed puberty.8,10
Care should be taken to distinguish GD from conditions that present with similar features, such as other lipid storage disorders (e.g. Niemann–Pick disease, Tay–Sachs disease and Pompe disease), haematological malignancies (e.g. leukaemia, lymphoma and multiple myeloma), immune thrombocytopenic purpura, autoimmune disease, hepatic cirrhosis, idiopathic avascular necrosis, viral disease, idiopathic splenomegaly and anaemia of chronic disease.10,31 The presence of hepatomegaly and polyclonal gammopathy without neutropenia can be useful for distinguishing GD from haematological malignancies. However, malignancy and GD sometimes co-exist and therefore further tests are required.
Diagnostic tests
β-glucosidase activity tests
A definitive diagnosis of GD can be obtained by measuring β-glucosidase activity in peripheral blood leucocytes (normal range: 2.1–5.3 μmol/l/h33).4,8,10 However, this test is only conducted at specialised centres, and diagnosis is often delayed by large distances between test centres and hospitals. Although blood samples can be couriered to test centres, they must be analysed almost immediately. This is not always possible without prior booking. Consequently, the β-glucosidase assay might not be performed during the initial visit.
To accelerate the diagnostic process, two cost-effective, non-invasive DBS assays have been developed for screening β-glucosidase activity.35 The benefit of these assays is that samples can be posted to a test centre via regular mail and stored until required. Both the tandem mass spectroscopy (MS/MS) DBS assay36 and the fluorescence DBS assay37 utilise a synthetic substrate instead of the native substrate, and both are associated with a low risk of false negative results. However, the fluorescence assay yields a relatively high rate of false positives;35,38 consequently, positive results must be followed up using the traditional assay.
Mutation analysis
Molecular screening for common GBA1 mutations is a cheap, reliable test for Ashkenazi Jews and other individuals who are at high risk for GD.4,8,10 In other populations, this technique may be complicated by a number of factors:
- It is not always possible to distinguish mutations in the active gene from those in the highly homologous pseudogene.
- A negative screen for common mutations does not exclude GD.
- The correlation between genotype and phenotype is weak.
Bone marrow biopsy
Bone marrow biopsy is useful for distinguishing haematological malignancy from GD, but should not be the first choice in Ashkenazi Jews and is rarely recommended in children due to the invasive nature of the test.4,8,10 Moreover, the presence of Gaucher cells in bone marrow (Figure 1) is not necessarily diagnostic of GD, because ‘pseudo’ Gaucher cells are often associated with other conditions (Table 1).39 Conversely, failure to detect Gaucher cells in a bone marrow aspirate cannot be used to exclude GD.

Biochemical markers
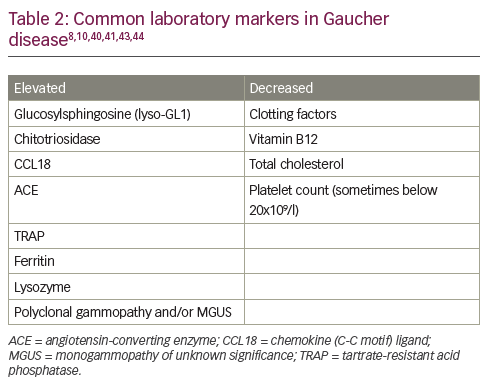
Abnormal levels of one or more biochemical markers (Table 2) are suggestive of GD; however, results are generally not sufficient to diagnose GD because similar changes can be caused by other disorders.40–42 The most reliable and specific biomarker for GD identified to date is glucosylsphingosine (lyso-GL1).43,44
Algorithms for the diagnosis of Gaucher disease type 1
Diagnostic algorithms designed to increase physician awareness of GD, prevent unnecessary biopsies and shorten the time from symptom development to diagnosis/management have been validated both in adults10 and in children.8 A diagnostic algorithm has also been developed for children suspected of having an LSD.45
Gaucher disease diagnosis in adults
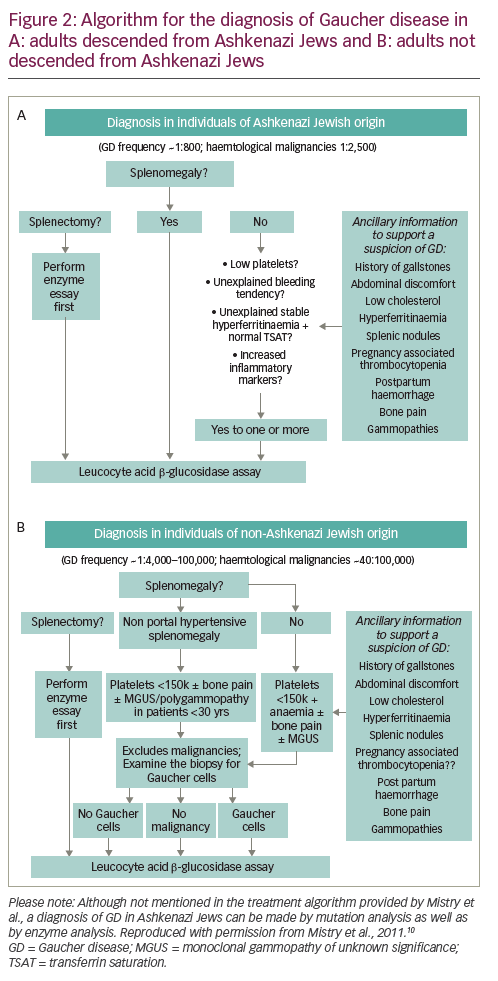
Given the relative incidence of GD type 1 versus haematological malignancy in the Ashkenazi Jewish population – approximately 1/800 versus 1/2,500 – algorithms for the diagnosis and management of GD in adults suggest that β-glucosidase activity should be routinely assessed in all high-risk individuals with thrombocytopenia and splenomegaly (Figure 2A).10 The GD diagnostic test should also be performed in patients without splenomegaly, but with one or more of the following: thrombocytopenia, bleeding tendency, unexplained stable hyperferritinaemia with normal TSAT and/or increased levels of inflammatory markers. Invasive tests, such as bone marrow biopsies, are not necessary and should be avoided in this population.
In people not descended from Ashkenazi Jews, splenomegaly is more likely to be caused by haematological malignancy than by GD (incidence 40/100,000 versus 1/40,000–100,000).10 In these patients, it is reasonable to perform a bone marrow biopsy before GD is considered to rule out malignancy (and other conditions associated with splenomegaly, such as portal hypertension due to liver diseases, haemoglobinopathies and chronic haemolytic anaemias) (Figure 2B). When interpreting biopsy results, it is important to remember that malignancy and GD sometimes co-exist,15 that Gaucher-like cells (Figure 1) are not exclusive to GD (Table 1) and that a diagnosis of GD should not be excluded if Gaucher cells are not found in patients with other signs/symptoms of GD.8
Although splenectomy is indicated for a number of conditions associated with splenomegaly, it tends to worsen the clinical course of GD in the liver, skeleton and the lungs.46 Consequently, GD must be excluded in all patients being considered for splenectomy. Assessment of splenomegaly should be combined with an examination of spleen parenchyma using abdominal ultrasound or magnetic resonance imaging. In patients with GD, the incidence of focal splenic defects caused by collections of Gaucher cells is approximately 20%, with incidence increasing with spleen size.47
Gaucher disease diagnosis in children/young adults
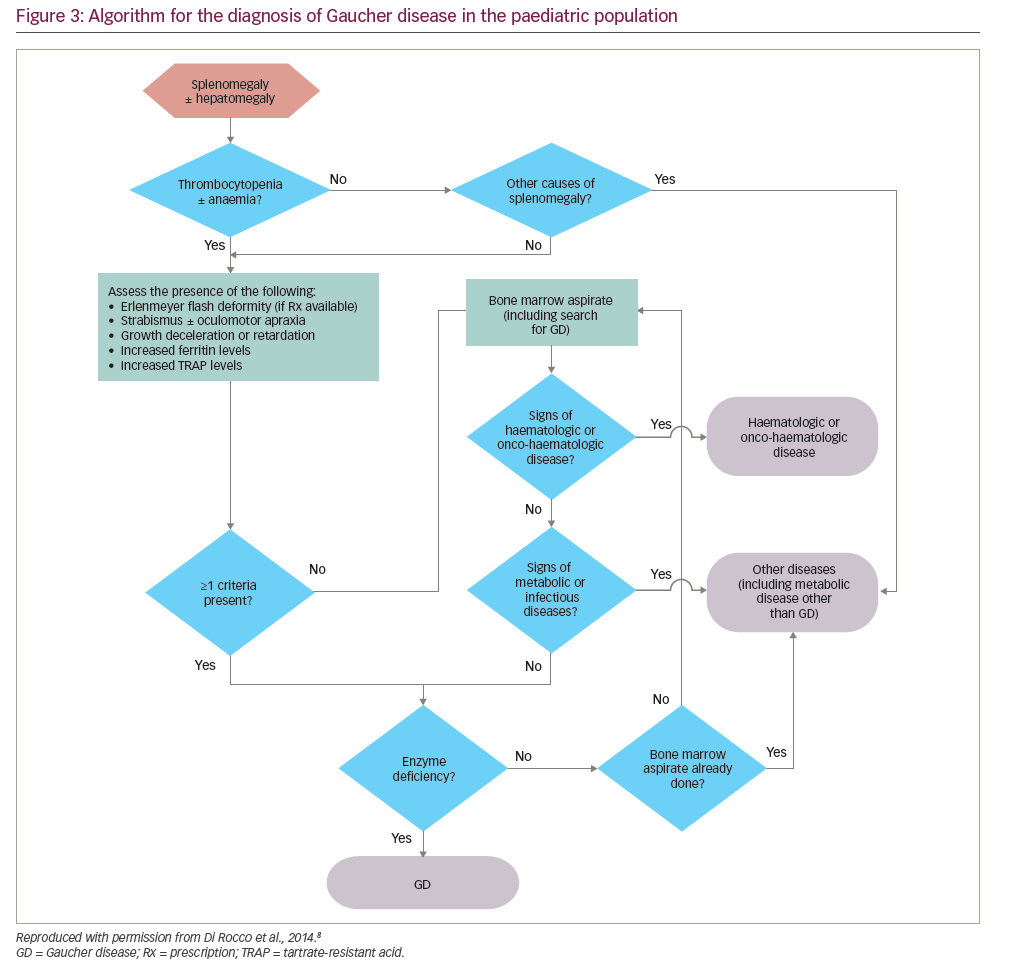
Algorithms for the diagnosis and management of GD (types 1 and 3) in children/young adults propose that GD should be considered in all children with unexplained splenomegaly, with or without thrombocytopenia, anaemia and/or hepatomegaly (Figure 3).8
Where possible, GD should be confirmed using a β-glucosidase assay, thereby avoiding unnecessary emotional trauma and/or bleeding that might be caused by bone marrow biopsy or liver biopsy.8 A DBS assay is recommended in children with unexplained splenomegaly, with or without hepatomegaly, who have thrombocytopenia and/or anaemia and at least one of the following:
- eye movement disorders (strabismus and/or oculomotor apraxia),48
- growth deceleration or retardation (occurs in 34% of children with GD),49
- the Erlenmeyer flask deformity (EFD; a skeletal deformity present in 49% of children with GD),49 and/or
- elevated levels of ferritin and tartrate-resistant acid phosphatase (TRAP).
Although bone pain is reported in 27% of patients with early GD,25 the subjective nature of pain excludes it as a reliable criterion.
In children with splenomegaly without thrombocytopenia or anaemia, a diagnosis of malignancy should be considered before testing for GD.8 If findings from a detailed medical history, physical examination and first-line laboratory tests do not suggest malignancy, an immediate enzyme assay is warranted. If malignancy is suggested, a bone marrow aspirate/biopsy should be performed. Metabolic diseases, including GD, should be considered in the absence of blood disorders, malignancy or infection (e.g. visceral leishmaniasis), even if Gaucher cells are not found in the aspirate.
In all cases, a diagnosis of GD should be followed by mutation analysis to determine the causative GBA1 mutation, thereby predicting the most likely GD type and the most appropriate management strategy.
Impact of implementing diagnostic algorithms and simple diagnostic tools
A survey of 16 medical experts from 14 GD specialist centres across 12 countries found that the main causes of diagnostic delay in patients with GD are a lack of awareness of GD/misdiagnosis (54% of experts), followed by phenotypic heterogeneity/non-specific symptoms (23%), mild symptomatology (15%) and outsourced testing (8%).32 Moreover, a survey of 406 haematologist–oncologists found that <50% of physicians were aware that the enzyme assay is the most appropriate diagnostic test for GD type 130 and a retrospective evaluation of 86 GD type 1 cases found that, despite clear guidelines discouraging the use of invasive procedures in people with suspected GD, the majority of patients had been diagnosed via bone marrow biopsy.31 It therefore follows that the implementation of simple diagnostic algorithms and the development of rapid diagnostic tools, such as the DBS assay, will avoid the need for unnecessary biopsies, reduce diagnostic delays and enable prompt (more effective) GD management.
To test this hypothesis, a study conducted in 35 haematology outpatient units across Italy was designed to estimate the prevalence of GD type 1 among 196 non-Ashkenazi Jewish adults (61 females, 135 males; mean age 47.8 ± 18.2 years) with splenomegaly and/or thrombocytopenia, diagnosed using the algorithm described by Mistry et al. (Figure 2).10 Patients were tested for β-glucosidase enzyme activity using the fluorescence-based DBS assay and, where possible, positive results were confirmed using the traditional assay and molecular GBA1 analysis. Overall, 34 of the 196 patients had a DBS value <4.4 pmol/punch/h (17.35%), and one patient had a negative DBS result despite a family history of GD. Overall, 7 patients (3.6% [95% confidence interval: 1.4–7.2] of the overall population; 1/28 patients) tested positive for GD using the β-glucoside activity assay, 10 (5.1%) were not available for follow up and 18 (9%) were GD-negative. To put these results into context, an initial survey showed that 18% of patients attending the same clinics each year had splenomegaly and/or thrombocytopenia, and that, using traditional diagnostic procedures, 11% of this subgroup never received a diagnosis. This suggests that the combined use of a diagnostic algorithm and DBS assay facilitates GD diagnosis and might reduce diagnostic delays among adults with splenomegaly and/or thrombocytopenia. A similar study – the GAUcher disease in Paediatric patients (GAU‐Ped) study – is ongoing in 53 paediatric haematology and oncology centres across Italy, using the diagnostic algorithm developed for children/young adults and the fluorescence-based DBS assay.8
Summary
Early diagnosis and prompt management of GD has the potential to reduce the risk of long-term GD complications while reversing many of the initial signs/symptoms. Most patients with GD have unexplained splenomegaly and/or thrombocytopenia, and many present during childhood; consequently, the majority of initial referrals are to haematologists and/or paediatricians.32 These physicians often fail to consider GD in their differential diagnosis due to a lack of awareness of rare haematological disorders, phenotypic heterogeneity and/or mild or non-specific symptoms.31 Diagnostic delays have also been attributed to expensive, time-consuming, outsourced testing.
Studies conducted in adults across Italy suggest that the use of simple diagnostic algorithms and newly-available DBS assays facilitate the diagnosis of GD and avoid unnecessary biopsies, even among physicians without disease-specific expertise.42 Although not definitive, evidence suggests that this may lead to a reduction in the diagnostic delay, thereby enabling prompt management and subsequent improvements in both clinical outcome and quality of life. Greater efforts are needed to raise awareness about the benefits of prompt GD diagnosis and management among non-GD experts.9