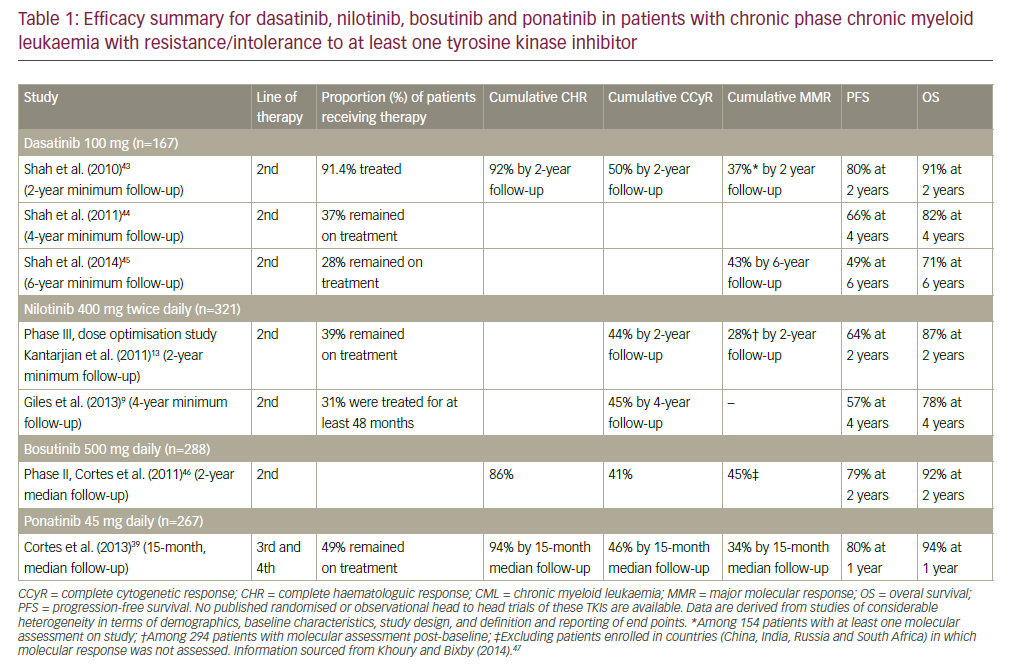
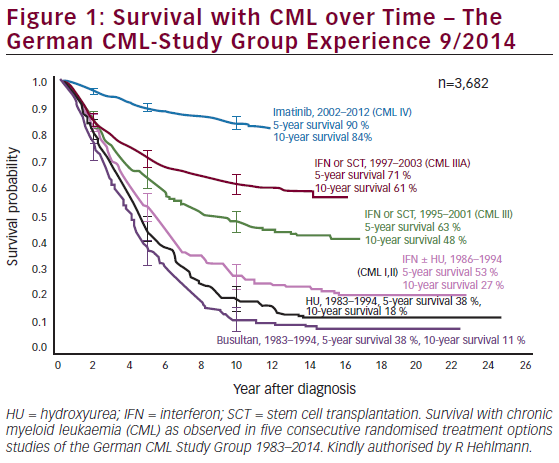
Unfortunately, AML patients classified as high-risk due to an unfavorable karyotype or secondary leukemia respond poorly to this chemotherapy.1 For these latter patients, the induction of complete remission is <50% with <10% relapse-free survival after post-remission therapy with high-dose ARA-C.1,2 There is an urgent need to improve the effectiveness of chemotherapy for these high-risk patients. For AML patients with a favorable prognosis, the major weakness of conventional chemotherapy is the maintenance of patients in CR. Even though post-remission therapy using high-dose ARA-C has increased survival of patients with AML, the optimal dose-schedule of this form of intensive chemotherapy is still unknown.1,3 About 60–70% of these latter patients will relapse after high-dose ARA-C post-remission therapy and die from disease progression. Despite the improvements in standard chemotherapy and supportive care, we need more effective chemotherapeutic agents to improve the survival of patients with AML.2
Epigenetic Therapy
Past research has shown that genetic changes, such as mutations and chromosomal aberrations and translocations, are important factors in the etiology of AML. Recent research has discovered that epigenetic changes, such as DNA methylation, can also play a key role in the development of leukemia.4 Epigenetics is defined as an inheritable change in gene expression that is not due to changes in the sequence of DNA.5 Gene silencing can occur by DNA methylation, which involves the enzymatic conversion of cytosine to 5-methylcytosine in the promoter region of genes. The normal function of this epigenetic event is to program the cell to silence the genes that are not essential for the function of each specific cell type. However, aberrant DNA methylation that silences tumor suppressor genes can also lead to the development of malignant disease.5 This epigenetic change can occur early in normal stem cells5 and subsequent transformation to leukemic stem cells can take place.7 An aberrant methylation pattern, such as promoter hypermethylation resulting in gene silencing, is one of the most common alterations in leukemia.4 Many loci in AML cells show aberrant DNA methylation.8,9 Since epigenetic events are reversible, they can be targets for chemotherapeutic intervention. An interesting epigenetic drug to use for this purpose is 5-aza-2’-deoxycytidine (dacogen, decitabine, DAC). DAC is a potent inhibitor of DNA methylation, which can reactivate tumor suppressor genes silenced by DNA methylation (see Figure 1). DAC can induce in vitro differentiation and loss of clonogenicity of human leukemic cells.10 DAC is an S-phase specific agent with a short in vivo half-life. Like ARA-C, its pharmacological activity is dependent on the dose schedule. The first clinical studies on DAC showed that this analog could induce complete remissions with advanced acute leukemia.17 In May 2006, DAC was approved by the FDA for the treatment of myelodysplastic syndrome (MDS) and currently is in clinical trial for the treatment of patients with AML who fail on conventional chemotherapy. Pre-clinical studies have shown that DAC can reactivate silent p15 and other tumor suppressor genes in AML cells.9 Pre-clinical in vitro and in vivo studies indicate that DAC is a more potent antineoplastic agent compared with ARA-C. At equimolar concentrations, DAC produced a greater loss of clonogenicity of human myeloid leukemic cells than ARA-C.11 In a mouse model bearing L1210 leukemia at maximum tolerated doses, DAC was able to cure 100% of mice, but no cures were observed with ARA-C.12 These preclinical studies showed that DAC has a higher therapeutic index than ARA-C.Optimization of DAC Dose Schedule for Acute Myeloid Leukemia Therapy
The important question is whether or not DAC will be more effective than ARA-C for the treatment of AML. Another important question is the optimal dose schedule of DAC for the treatment of patients with AML. The current recommended dose schedule of DAC for the treatment of MDS is 20mg/m2 (one hour IV infusion) daily for five days.13 This low dose schedule of DAC is well tolerated in patients >60 years or with a very poor hematological status. About >80% of MDS patients will relapse after low-dose DAC therapy. The duration of second disease remissions for these MDS patients was inferior to the first treatment with DAC.14 These observations suggest that low-dose DAC can permit some recovery by leukemic stem cells, possibly due to remethylation of tumor suppressor genes.6 According to pre-clinical data, at higher doses of DAC these changes are irreversible, resulting in a complete loss of the clonogenic potential of the leukemic stem cells. The success with high-dose ARA-C for the therapy of AML suggests that a more intensive dose schedule of DAC could be used for the treatment of these patients. The pharmacology of ARA-C and DAC is almost identical for these two deoxycytidine analogs. Both these S-phase specific agents are phosphorylated by the same enzyme, deoxycytidine kinase, and incorporated into DNA. However, their mechanisms of action are different. ARA-C acts as a chain terminator, producing a potent inhibition of DNA synthesis, whereas DAC is a potent inhibitor of DNA methylation.12 The early published data on ARA-C can be used to help design the optimal schedule for DAC.15 The initial phase I studies on ARA-C showed that a continuous infusion was a more effective schedule than daily injections in the treatment of patients with acute leukemia.16 What would be the optimal duration of the infusion of DAC? For conventional doses of ARA-C, a 120h infusion was reported to be more effective than 48h infusion.16 The optimal schedule for DAC may be somewhere between 48 and 120h.15
The major side effect produced by DAC is its hematopoietic toxicity. If one assumes that ARA-C and DAC produce a similar pattern of hematopoietic toxicity, it may be possible to use intensive DAC therapy without encountering unacceptable toxicity.15 Both these analogs permit the survival of resting hematopoietic stem cells (i.e. cells not in S-phase), which allows for the recovery from myelosuppression after intensive-dose therapy. The doseschedules of ARA-C can be used as a guideline for dose-schedule selection for initial clinical trials for DAC. At total dose of 1,080–2,400mg/m2 as continuous infusion for 36–40h, DAC produced similar myelosuppression17 to that reported for a similar schedule for ARA-C in adults.18 Furthermore, most leukemic patients in relapse are able to recover from the myelosuppression produced by six days (144h) of high-dose ARA-C therapy.19 The selection of ideal AML patients for experimental chemotherapy with DAC is a challenge. Since induction with conventional dose ARA-C followed by post-remission therapy with high-dose ARA-C can produce >50% longterm survivors for low-risk AML, this group of patients are not candidates for DAC therapy. The ideal candidates for experimental therapy are the high-risk AML patients, since they respond poorly to conventional chemotherapy and have a very short life expectancy. This latter group consists of patients with adverse cytogenetics, with MDS that progressed to AML and secondary AML.
In a pilot clinical trial, we administered DAC at 30mg/m2/h for 60h to patients with very advanced AML and obtained some complete remissions.15 However, the remissions for these patients were short, possibly due to the fact that the duration of therapy was not long enough to permit all of the leukemic cells to progress and enter the S-phase, the phase of the cell cycle in which DAC exerts its antileukemic action. For initial clinical trials, we suggest the following dose schedule: DAC at 30mg/m2/h for three days (72h). This DAC dose schedule can be modified depending on the observed response and toxicity. For example, the duration of the IV infusion can be increased by increments of 12h while maintaining the dose rate constant. The dose-limiting toxicity produced by DAC is myelosuppression.Conclusion
In conclusion, the full chemotherapeutic potential of epigenetic therapy of AML is unknown. DAC has a fascinating mechanism of action of reactivating silent genes that suppress leukemia progression. This action of DAC can induce the leukemic stem cells to undergo differentiation or senescence resulting in an irreversible loss of their proliferative potential. The pre-clinical studies indicate that DAC is a more potent antileukemic agent than ARA-C. These observations provide the rationale to support the initiation of clinical trials to optimize the dose schedule of DAC for patients with high-risk AML, who have very little chance of survival with the current conventional chemotherapy.