Pancreatic ductal adenocarcinoma (PDA) is an aggressive disease with a persistently high mortality that has only marginally decreased in recent decades.1–4 The disease typically presents with nonspecific symptoms in the early stages, often leading to delayed diagnosis and a poor prognosis. Approximately 80% of patients with PDA present at a late stage.5,6 Consequently, most patients have advanced disease (stages III or IV) at diagnosis that is beyond curative resection.7–9 As there is a lack of reliable blood borne or other biomarkers for PDA, early diagnosis is difficult.10 Effective non-surgical treatment for all stages of PDA continues to be an urgent unmet medical need.1,8 Improved understanding of the disease is yielding new approaches to treatment which may lead to better prospects in the future for managing PDA.8 Here, we summarize the prevalence, pathophysiology, diagnosis and current standards of care and discuss novel treatment options in clinical development for PDA.
Pancreatic cancer demographics
PDA represents over 90% of cancers arising in the pancreas and carries an extremely poor prognosis.8 PDA is the third leading cause of cancer death in the US, the fourth leading cause in Europe and the fourth or fifth in Asian nations.11–13 It is projected to become the second leading cause of cancer-related death in the USA by 2030.14 Mortality rates vary throughout the world according to GLOBOCAN 2012 estimates, with 6.9/100,000 in the USA, 6.8/100,000 in Western Europe, decreasing to less than 1.0/100,000 in Middle Africa and South Central Asia.15 The National Cancer Institute’s estimates for new diagnoses of pancreatic cancer in the USA in 2018 is 55,440 (12.6/100,000, 3.2% of all new cancer cases) with 44,300 deaths (10.9/100,000, accounting for 7.3% of all cancer deaths).11 The estimated prevalence in the USA in 2015 was 68,615 (17.1/100,000).11 In Europe, the incidence in 2018 is estimated as 132,559 (17.8/100,000) with 128,045 deaths (17.2/100,000). The incidence in Asia in 2018 is estimated as 214,499 (4.7/100,000) with 200,681 deaths (4.4/100,000).16
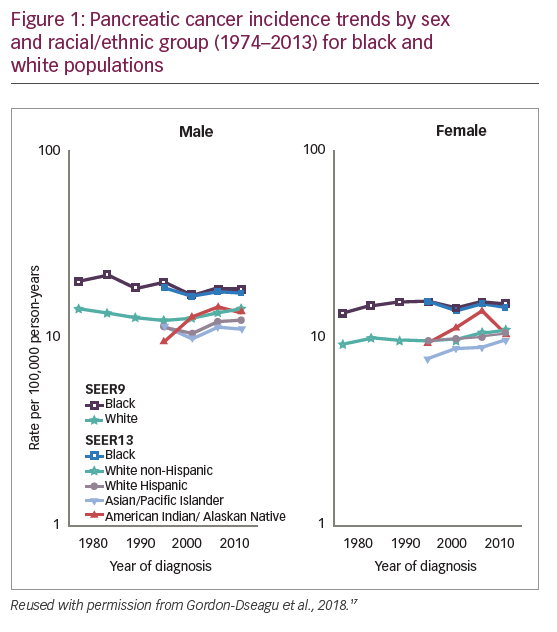
Data from the U.S. Surveillance Epidemiology and End Results registries show that among white males, the incidence of pancreatic cancer decreased from 1975 to 1994 (annual percentage change [APC]: -0.98%) but increased during 1994–2013 (APC: +0.95%).17 Among non-Hispanic white and Hispanic males, the incidence APC between 1992 and 2013 was +0.84% and +0.73%, respectively. Incidence during this period also increased in white non-Hispanic, Hispanic and Asian females (APC: +0.81%, +0.56% and +1.23%, respectively), and even more rapidly among females aged 25–34 years (APC >+2.5%). Incidence among black males and females remained unchanged, but was higher than amongst white, Hispanic and Asian populations (Figure 1).17
The PDA mortality rate is remarkably similar to incidence; survival rates have only improved slightly over the past 40 years.11 According to EUROCARE-5, the fifth cycle of the EUROCARE study of cancer survival in Europe, the 1-, 3-, and 5-year survival rates of patients with pancreatic cancer diagnosed during 1999–2007 were only 26%, 9%, and 7%, respectively.18 More recent data from a study of European and USA patients diagnosed during 2003–2014 shows little change in this figure, with a 1-year survival of 19–34% and 5-year survival of 4–11%.19 Survival of patients with stages III (locally advanced) and IV (distant metastatic) PDA was much worse than for stage I–II patients with potentially resectable disease. Median overall survival (OS) for stage III and IV PDA was <1 year and <6 months, with a 5-year survival of only ~2%.19 By comparison, the 5-year survival rates for other common cancers are much higher: prostate cancer 98.2%, breast cancer 89.7%, colorectal cancer 64.5%. Even outcomes from lung cancer (5-year survival 18.6%)11 exceed those for PDA, reflecting introduction of effective treatments over the last decade.
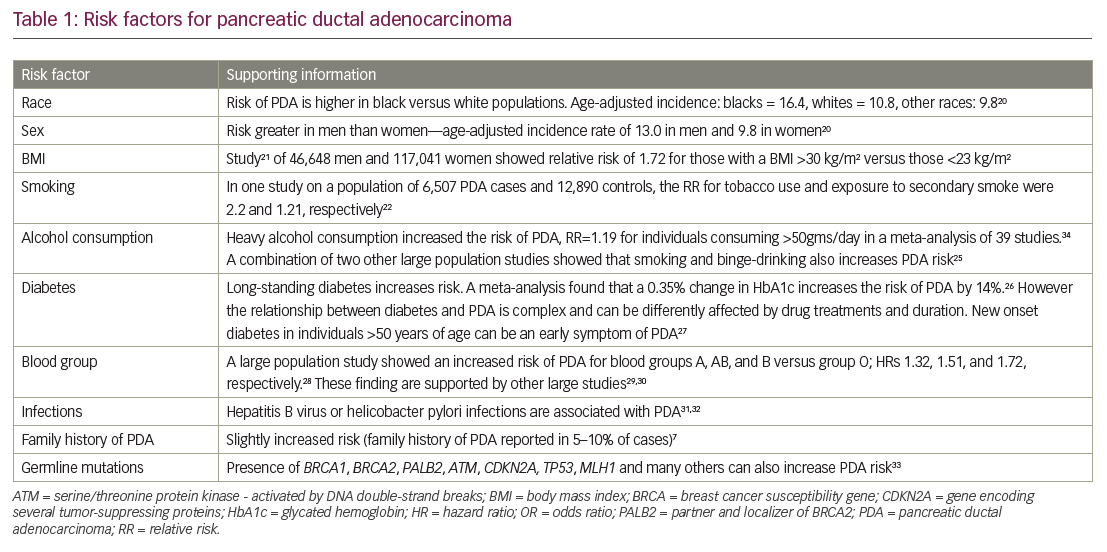
Several different risk factors for PDA have been identified: there are associations with lifestyle (e.g. smoking, obesity, and alcohol consumption), infection and disease history, as well as genetic factors, blood group, and family history (Table 1).7,20–34 Type 2 diabetes (T2D) is frequently associated with PDA,20,26,27,35,36 although the nature of the relationship is not clear. Recent-onset T2D appears to be a manifestation of PDA whereas long-term T2D may be a risk factor for PDA.36
Pathophysiology and natural history of pancreatic cancer
Approximately 95% of all pancreatic cancers are exocrine adenocarcinomas, most of which originate in the ducts and at the head end of the pancreas.35 Evidence suggests that it can take many years for an initiating abnormal pancreatic cell to become malignant.37 However, PDA may progress rapidly through the clinical stages; a study of 13,131 patients calculated that the average T1-stage PDA advances to T4 stage in just over a year.38 The tumor arises from ductal cells via pancreatic intraepithelial neoplasia (PanIN) precursor lesions (Figure 2).39
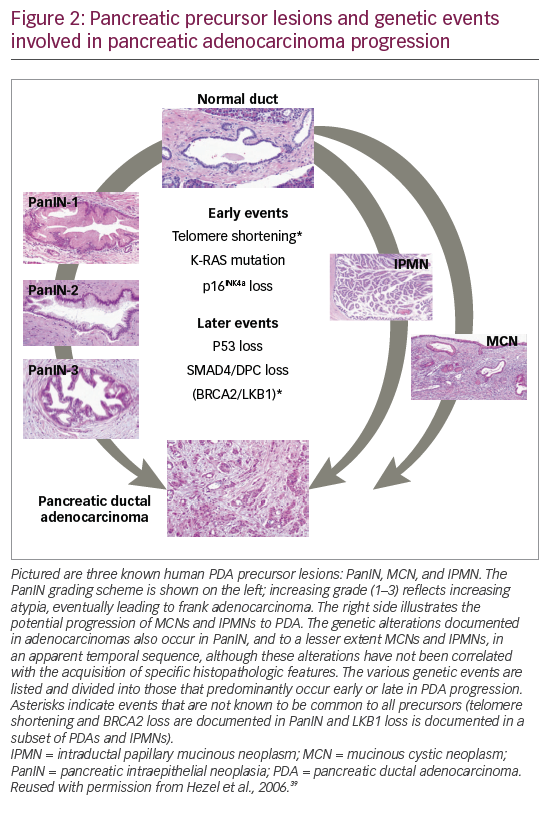
Epidemiologic studies have shown that 5–10% of PDAs are associated with an inherited component.40 A recent case-control study found that 5.5% of patients with PDA (167/3,030) had deleterious mutations in six genes which were associated with a predisposition to PDA (CDKN2A, TP53, MLH1, BRCA2, ATM, and BRCA1). Among all tested patients in this study, 7.9% (27/343) with a family history of PDA and 5.2% (140/2,687) with no family history of PDA had a mutation in one of these six genes.33
The most frequent somatic alterations in PDA are in the KRAS, CDKN2A, TP53, and SMAD4 genes.41,42 KRAS alterations are observed in >90% of PDAs and occur early in tumor development.42 In addition to these prevalent gene alterations, a “long tail” of gene mutations is observed, including those involved in axon guidance and DNA repair.43–45 These mutations often alter the metabolism of pancreatic tumors. PDA cells also have a notable genomic instability phenotype that enables the rapid development of treatment resistance.9
Molecular analysis of metastatic PDA tumors using real-time genomic characterization can identify clinically relevant alterations that can potentially guide treatment of PDA and the development of new treatment (Figure 3).42–44,46 Studies by the Australian Pancreatic Cancer Genome Consortium on whole genomes in 100 different PDAs found that chromosomal rearrangements could be classified as stable (20%), scattered (36%), unstable (14%), and locally rearranged (30%) and these types affect response to treatment. The study also identified a mutational signature of DNA damage repair deficiency, which may be useful in predicting therapeutic response.47 Defining patients with hypermutation, i.e., a high mutation burden, may help predict a patient’s response to immunotherapy.48 Next-generation sequencing can identify mismatch repair deficiency, which is also predictive of tumor response to immunotherapy.49 Up to 7% of PDAs have loss-of-function mutations in either BRCA1, BRCA2, or both BRCA genes and germline BRCA mutation may be a biomarker for response to poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors.50
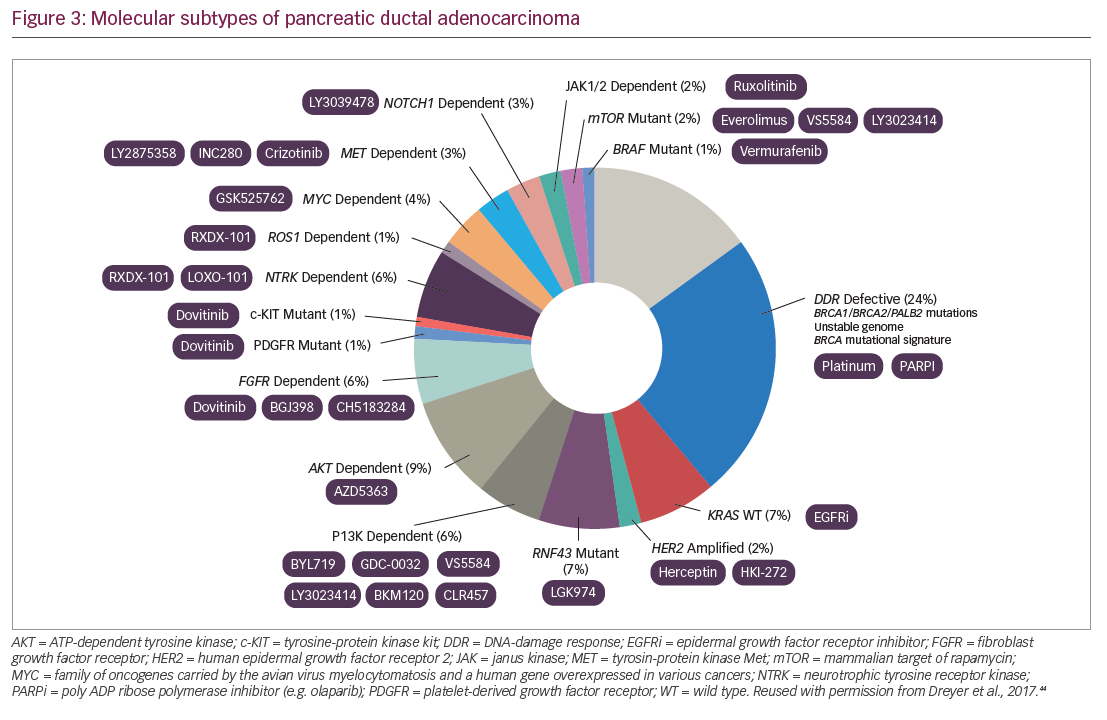
Tumor genotyping and gene expression assessments are likely to become increasingly useful in defining which patients are more likely to respond to chemotherapy and immunotherapy.51,52 The US Pancreatic Cancer Action Network ‘Know your Tumor®’53 initiative allows patients to receive molecular profiling of their tumor to facilitate a personalized approach. In this initiative, patients have undergone multiomic profiling involving genomic, proteomic, and phosphoprotein-based analysis. Initial findings from 640 participating patients showed highly actionable findings in over 25% of tumors. Among these patients, those who had received matched therapy had significantly longer progression-free survival (PFS) than those who received unmatched therapy (4 months versus 2 months; hazard ratio [HR] 0.47; adjusted p=0.03). These findings indicated that molecular profiling has considerable potential in PDA diagnosis and treatment selection.52
In addition to genomic alterations, other fundamental features of PDA have started to guide ongoing research efforts. In PDA, the tumor microenvironment is created by interactions between cells such as pancreatic epithelial cells, cancer-associated fibroblasts, stellate cells, adipocytes, and infiltrating immune cells. Such interactions are believed to promote tumor growth and metastasis (Figure 4) and may contribute to resistance to systemic therapy.54–56 Stellate cells and adipocytes in the tumor microenvironment are believed to act synergistically to supply the cancer cells with metabolites such as glutamate and other micronutrients.57,58 These cells also maintain an inflammatory state which is accompanied by the infiltration of the tumor by inflammatory cells such as lymphocytes, neutrophils and macrophages.59,60 Metabolism is altered within PDA cells and this is affected by the stroma which limits perfusion of the tumor tissues.61,62 The stroma surrounding PDA tumors is rich in hyaluronan which has been implicated in increasing intertumoral pressure, collapsing blood vessels, and preventing access by therapeutic agents and immune cells.63,64 The stroma may also promote metastasis, with the possibility that tumors having a high-extracellular hyaluronan level (hyaluronan-high, defined as hyaluronan staining in the extracellular matrix ≥50% of the entire tumor surface at any intensity) may be more aggressive than tumors with low hyaluronan (Figure 4).54,55,65–67 Overcoming this barrier using enzymatic depletion of hyaluronan is an important development in current treatment approaches.66,68
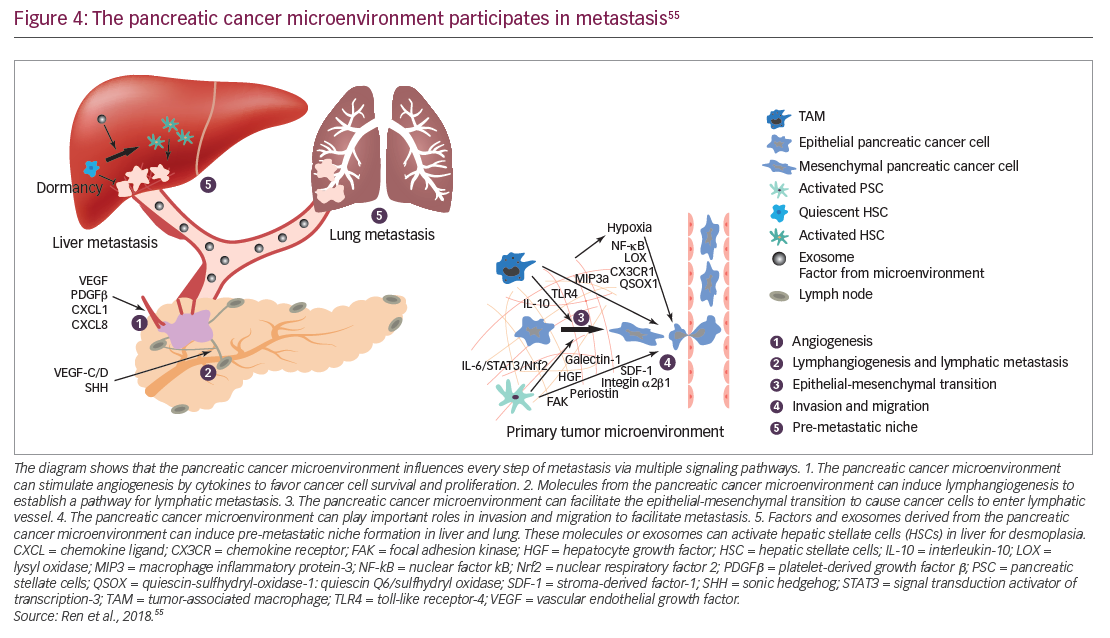
Within PDAs, cancer stem cells have an important role and have unique metabolic, autophagic, invasive, and chemoresistance properties, enabling continuous self-renewal and allowing the cells to evade elimination from chemotherapies. They also promote metastases.69 Current treatment options do not target cancer stem cells, but improved understanding of the characteristics and signals that maintain and drive this cancer stem cell population is vital for the development of more effective treatments.
Diagnosis
Symptoms of PDA include weight loss, jaundice, loss of appetite, malabsorption, severe pain in mid abdomen and back, light-colored stools or dark urine, dyspepsia, fatigue, and nausea.70 In many cases, however, the disease creates mild or non-specific symptoms during its early stages, and as a result, appropriate investigation is delayed and <20% of pancreatic cancers are amenable to complete surgical resection at diagnosis.9,71 In advanced PDA, symptoms such as pain and thromboembolic events are highly prevalent72–74 and are associated with increased morbidity and mortality.72 In addition, cachexia develops in approximately 80% of patients with PDA during their disease course, and up to one-third of patients die from complications from cachexia.75 Diagnosis of PDA involves multiple imaging techniques. Initial investigations of abdominal symptoms may include ultrasound and computed tomography (CT) examinations. Endoscopic ultrasound is the optimal modality to assess and stage primary tumors as well as to obtain a tissue diagnosis.76 Positron emission tomography is potentially of value in improving selection of patients for surgical resection.77
Detecting PDA earlier would be advantageous but there are no reliable diagnostic circulating biomarkers for the disease.10 Serum CA 19-9 is widely used as a marker for metastatic PDA, but is non-specific and is not recommended for diagnostic use.78 Serum macrophage inhibitory cytokine 1 (MIC-1) also has some potential as a biomarker, especially in combination with CA 19-9.79 A recent study in a large cohort of patients with PDA found that a signature of 29 biomarkers can reliably discriminate individuals with stage I and II PDA from control subjects and this was validated in a further prospective study at a separate treatment center.80 A combined analysis of a panel of serum proteins and microRNAs showed improved sensitivity and specificity in the detection of PDA-initiating cells and other biomarkers in patients with PDA compared with healthy controls but further development of the technique in a larger population is needed.81,82
In the general population, widespread screening is not feasible at present due to the lack of suitable biomarkers and the comparative rarity of the disease. Surveillance, however, is warranted in individuals >50 years of age with one first-degree relative, two family members who are affected by PDA, or in those carrying a mutation in a known cancer predisposition gene.33,83,84 Recent results from a long-term (16 years) surveillance study indicated that 9 of 10 PDAs detected during surveillance were resectable and 85% of these patients survived for 3 years, suggesting a benefit of surveillance in high-risk PDA.85
Current standards of care and their efficacy
Current treatment in PDA is based on disease stage at diagnosis (Table 2).86 Given the relative recalcitrance of PDA to current treatments, the National Comprehensive Cancer Network in the USA indicates that clinical trials are a preferred treatment option at every stage of pancreatic cancer diagnosis.

Potentially resectable primary pancreatic ductal adenocarcinoma (stages I, II, and III)
Stage I and II PDAs, and stage III PDAs that are not invading local vessels, are potentially resectable. The standard operation for tumors of the pancreatic head is the pancreaticoduodenectomy (Whipple procedure); tumors of the body or tail are resected using distal pancreatectomy.87 Surgery is the only means of curing PDA and radical surgery achieving R0 resections offers the best outcomes when combined with optimal adjuvant chemotherapy.88 Outcomes are less favorable for those patients with larger tumors and involved surgical margins, and they represent the majority of patients undergoing surgery.88 Therefore, the overall median survival for all patients undergoing resection of PDA has been reported at 18–27 months, and the 5-year survival rate ranges from 12–35%.89–91 Around 30% of patients undergoing resection may die within 1 year of surgery,92 indicating that PDAs invade and metastasize early. Since patients with involved resection margins have much poorer survival,93 patient selection for surgery is critical to optimize outcomes and avoid morbidity associated with major surgery. Development of novel strategies of perioperative adjunctive therapy is essential to reduce recurrence after surgery.
Following surgical resection of PDA, adjuvant chemotherapy can substantially prolong OS compared to surgery alone. In 2004, the European Study Group for Pancreatic Cancer-1 (ESPAC-1) trial demonstrated that adjuvant chemotherapy with 5-fluorouracil achieved 5-year OS of 21%, versus 8% for with surgery alone (p=0.009).94 Subsequent studies confirmed gemcitabine was as effective as fluorouracil as an adjuvant therapy and better tolerated.95,96 In 2017, the ESPAC-4 study results concluded that the addition of capecitabine to gemcitabine further improved outcomes, with estimated 5-year OS reaching 28.8%. Median OS was 28.0 months for patients receiving adjuvant gemcitabine + capecitabine and 25.5 months for those receiving gemcitabine alone (HR 0.82; p=0.032).88
Initial results from the phase III PRODIGE 24/CCTG PA.6 trial showed improved efficacy for modified fluorouracil and folinic acid, oxaliplatin, leucovorin and irinotecan (FOLFIRINOX) compared with gemcitabine. After a median follow-up of 30.5 months, the median disease-free survival for modified FOLFIRINOX versus gemcitabine was longer (22 versus 13 months), as was the median OS (54 versus 35 months).97 As in the metastatic setting, combination chemotherapy appears superior to single-agent gemcitabine in terms of efficacy, but careful patient selection is needed in view of greater associated toxicity. In addition, as nab-paclitaxel + gemcitabine demonstrated a clinically significant survival benefit versus gemcitabine alone in metastatic disease, it is also under investigation in patients with surgically resected PDA (APACT trial; NCT01964430).98 A total of 866 patients have been randomized, and primary results of disease-free survival and safety will be available at the American Society of Clinical Oncology (ASCO) 2019 annual meeting. However, according to the recent press release by the sponsoring company, the study did not meet the primary endpoint of disease-free survival by blinded independent review; although the secondary endpoint of OS at the time of this data cut off was improved, reaching nominal statistical significance.99,100 The safety profile observed in the study was consistent with previously reported studies of nab-paclitaxel.
The population with early PDA is a heterogeneous patient group and it is now clear that not all stage II tumors are actually resectable. Improvements in multidetector CT scanning with protocols optimized for PDA imaging offer higher resolution images of the tumor vessel interface, which has enabled evaluation of the extent of abutment and encasement of adjacent vessels. Tumors with no arterial contact are resectable, tumors abutting major pancreatic arteries ≤180o are termed “borderline resectable”, and tumors encasing major pancreatic arteries ≥180o are unresectable (Table 3, Figure 5).93
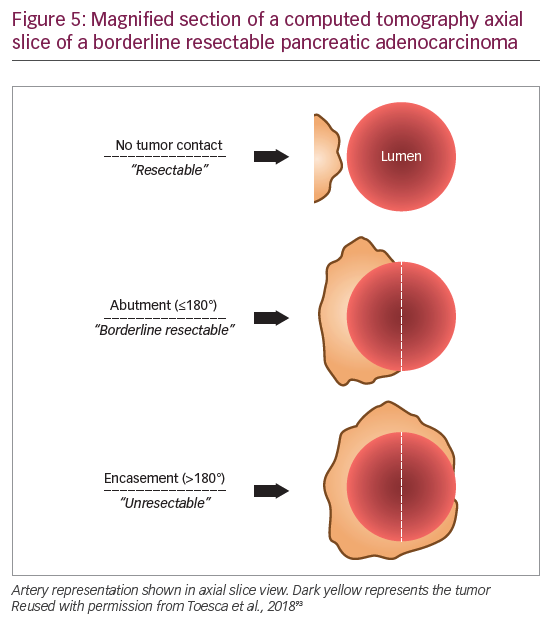
The significance of refining PDA resectability has been the rationale for neoadjuvant therapy aimed at improving outcomes from primary cancer surgery. One of the first major reviews evaluating this approach suggested that as many as one-third of unresectable tumors could be rendered resectable by neoadjuvant therapy.101 A 2017 propensity score-matched analysis of 15,237 stage I/II patients from the National Cancer Database, found that patients who received neoadjuvant therapy followed by resection had improved survival compared with those who received upfront resection (median survival, 26 months versus 21 months, respectively; HR 0.72; 95% confidence interval [CI], 0.68–0.78).102 A single-arm study presented at ASCO 2018 found that preoperative FOLFIRINOX chemotherapy was feasible in patients with resectable PDA and allowed for better selection of patients with more advanced tumors who were not initially suitable for resection,103 and a randomized trial of 364 patients presented at ASCO-GI 2019 found that neoadjuvant chemotherapy using gemcitabine and S1 resulted in improved OS compared to those receiving upfront surgery (median OS 36.7 versus 26.6 months, respectively; HR 0.72; 95% CI 0.55–0.94; p=0.015 stratified log-rank test).104
Neoadjuvant therapy is a promising exploratory approach, but a number of issues need to be addressed, including optimal treatment regimen and modalities. Additional randomized controlled trials in large patient populations are needed to properly evaluate this approach.
Locally advanced pancreatic ductal adenocarcinoma (unresectable stage III)
Diagnosis of locally advanced PDA confirms unresectability. Whether chemotherapy or chemoradiotherapy should be offered as treatment for this disease stage is an ongoing debate. However, combination systemic chemotherapy using in the setting of metastatic disease may be considered as initial therapy prior to chemoradiation for appropriate patients with locally advanced disease. The phase III LAP07 clinical trial found no significant difference in OS with chemoradiotherapy compared with chemotherapy alone. The same study found no significant survival advantage in patients with locally advanced PDA when erlotinib was added to gemcitabine as maintenance therapy.105
Results from the SCALOP phase II study showed that capecitabine was a more effective radiosensitizer compared with gemcitabine when used after induction chemotherapy, but the difference in the primary endpoint of PFS was not significant (15 months in the capecitabine group versus 13 months in the gemcitabine group; p=0.012).106 On the other hand, a meta-analysis of 11 studies involving 315 patients with locally advanced PDA reported that FOLFIRINOX chemotherapy was associated with a median OS of 24 months, which is superior to that achieved with single agent gemcitabine. The percentage of patients down-staged to enable surgical resection ranged from 0–43%, while the rate of serious or life-threatening adverse events associated with treatment reached 60%. The authors concluded that prospective randomized trials are needed to evaluate optimal chemotherapy and chemoradiotherapy in this patient group.107 In clinical practice, combination chemotherapy is being routinely offered to patients, with the option for consolidation chemoradiotherapy in those cases where surgical downstaging has not been achieved.108
Metastatic pancreatic ductal adenocarcinoma (stage IV)
Standard optimal first-line combination chemotherapy for metastatic PDA is based on the results of the MPACT (nab-paclitaxel + Gemcitabine in Metastatic Pancreatic Adenocarcinoma) and PRODIGE/ACCORD (FOLFIRINOX versus Gemcitabine as First-line Treatment for Metastatic Pancreatic Adenocarcinoma) trials.2,109,110 The MPACT trial showed that, in patients with metastatic PDA, nab-paclitaxel + gemcitabine significantly improved OS, PFS, and response rate, but rates of peripheral neuropathy and myelosuppression were increased. The median OS was 9 months in the nab-paclitaxel + gemcitabine group versus 7 months in the gemcitabine group (p<0.001).110 The PRODIGE/ACCORD study showed that first-line FOLFIRINOX produced a greater survival advantage in appropriately selected patients with metastatic PDA over gemcitabine (the median OS was 11 months in the FOLFIRINOX group versus 7 months in the gemcitabine group (p<0.001).109 Although FOLFIRINOX is now one of the preferred first-line options for patients with metastatic PDA, debate persists whether the survival benefits of the combination regimen in metastatic disease outweigh the associated toxicities. These include thrombocytopenia, febrile neutropenia, diarrhea, and neuropathy. Therefore, FOLFIRINOX is usually reserved for patients aged ≤76 years who have a good PS (ECOG 0 or 1).109 Modifications of the FOLFIRINOX regimen can provide comparative survival benefits to the conventional regime with fewer adverse events.111 Gemcitabine monotherapy remains an acceptable treatment option for those patients unable to tolerate combination regimens.
Second-line treatment may be offered to carefully selected patients, according to ECOG PS and other clinical factors, such as bilirubin levels and comorbidities. However, the advantage of second-line therapy is very limited and the optimum regimen not established.112 The German Charité Onkologie (CONKO) 003 phase III study found that second-line oxaliplatin + fluorouracil significantly extended OS compared with fluorouracil alone (6 months versus 3 months; p=0.01) in patients with advanced gemcitabine-refractory PDA. However, the oxaliplatin + fluorouracil group showed an increased rate of mild to moderate neurotoxicity (38.2% versus 7.1%).113 On the basis of these findings, fluorouracil/leucovorin + oxaliplatin was considered as the best option following failure of gemcitabine-based treatment. Conversely, the PANCREOX (Randomized Phase III Study of Fluorouracil/Leucovorin With or Without Oxaliplatin for Second-Line Advanced Pancreatic Cancer in Patients Who Have Received Gemcitabine-Based Chemotherapy) phase III study found that the addition of oxaliplatin to fluorouracil/leucovorin as modified FOLFOX6 (infusional fluorouracil, leucovorin, and oxaliplatin) had a detrimental effect compared with fluorouracil/leucovorin.114
The phase III NAPOLI-1 (nanoliposomal irinotecan with fluorouracil and folinic acid) study showed a survival advantage for nanoliposomal irinotecan fluorouracil + folinic acid compared with flurouracil/folinic acid (6 months versus 4 months; p=0.012) in patients with metastatic PDA after previous gemcitabine-based therapy.115 This represents another useful second-line treatment option.
Early evidence is emerging that subgroups of patients identified by a molecular signature may benefit from a targeted therapeutic approach. Up to 7% of PDAs harbour a germline BRCA mutation. BRCA genes code for proteins involved in homologous recombination repair of DNA double-strand breaks. Poly(adenosine diphosphate-ribose) polymerase (PARP) inhibitors which prevent repair of DNA single strand breaks have demonstrated antitumour efficacy in ovarian and breast cancer patients with a germline BRCA mutation. Very recently, the PARP inhibitor, olaparib, was reported to maintain response and delay disease progression after platinum-based chemotherapy in BRCA mutant advanced pancreatic cancer patients.50 Patients with BRCA-associated locally advanced PDA can benefit from targeted therapy with olaparib as a second-line therapy after treatment failure with another line of therapy.116
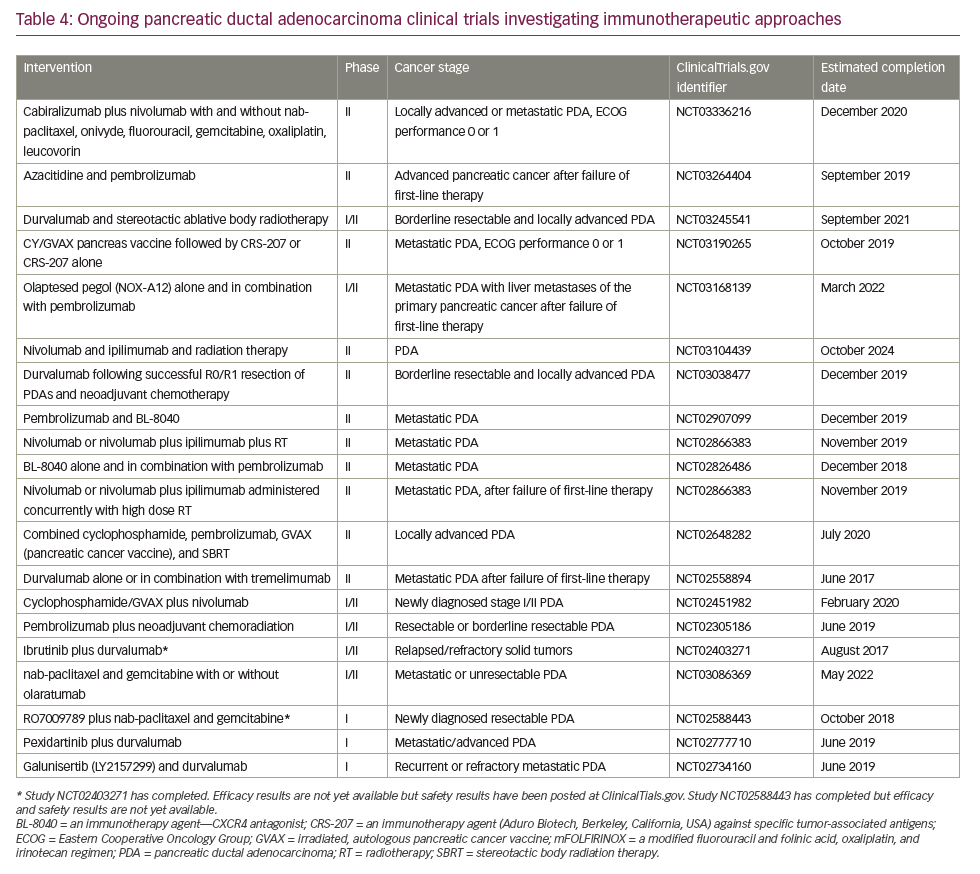
In May 2017, the anti-programmed cell death protein 1 (PD1) checkpoint inhibitor, pembrolizumab, was licensed in the USA to treat patients with DNA mismatch repair deficiency, determined either by genetic mutation analysis or immunohistochemical assessment of microsatellite instability. Researchers reported an objective response rate of 51% across 12 different tumor types and complete responses in 21% of patients.117 All patients were heavily pre-treated, having received two or more prior regimens. Mismatch repair deficiency of microsatellite instability is not routinely tested for in PDA and the license has not been extended outside the USA. However, these data are the first to identify a potential role for immune checkpoint inhibitors as treatment of PDA and many research studies are currently exploring novel immunotherapy approaches for this disease (Table 4). Further investigation of an immune checkpoint inhibitor combination for PDA treatment was conducted in a recent phase Ib/II study which investigated the use of the PD-L1 inhibitor durvalumab and the Bruton tyrosine kinase inhibitor ibrutinib given every two weeks to patients with pretreated solid tumours including 49 individuals with stage III/IV PDA.118 The results indicate that the combination was well tolerated but showed only limited anti-tumour activity in this study population.
According to the ASCO pancreatic cancer guidelines119 and a more recent update,120 the following options are updated or newly recommended for the treatment of metastatic PDA in 2018:
- For patients with Eastern Cooperative Oncology Group performance status (ECOG PS) ≥3, the major emphasis should be on optimizing best supportive care measures (evidence quality: intermediate).
- Routine testing for deficiency in mismatch repair or high microsatellite instability is recommended (evidence quality: low). For patients who tested positive for deficiency in mismatch repair or microsatellite instability, the checkpoint inhibitor such as pembrolizumab is recommended (evidence quality: intermediate).
- Gemcitabine + nab-paclitaxel can be given as second-line therapy to patients who have received first-line FOLFIRINOX and have an ECOG PS of 0–1 and a favorable comorbidity profile (evidence quality: low).
- Fluorouracil + nanoliposomal irinotecan, or fluorouracil + irinotecan as second-line therapy for patients who received first-line gemcitabine + nab-paclitaxel, have an ECOG PS of 0–1 and a favorable comorbidity profile (evidence quality: low).
- Fluorouracil + oxaliplatin may be considered as second-line therapy for patients who received first-line gemcitabine + nab-paclitaxel, have an ECOG PS of 0–1 and a favorable comorbidity profile (evidence quality: low).
- Gemcitabine or fluorouracil can be considered as second-line therapy for patients who have either an ECOG PS of 2 or a comorbidity profile that precludes more aggressive regimens and want to continue anticancer therapy (evidence quality low).
- No data available supporting third-line (subsequent-line) cytotoxic therapy.
Supportive care
PDA is a debilitating disease associated with multiple complex symptoms. In addition to anticancer therapies, patients diagnosed with PDA often need interventions to provide relief of biliary and/or duodenal obstruction, malnutrition, and pain.69 The endoscopic placement of self-expandable metal stents can be used to palliate biliary and duodenal obstruction.121 Patients with PDA are at high risk of thrombo-embolic events, which increase morbidity and mortality.71 Results from a randomized controlled trial indicate that anticoagulation may be useful for preventing serious morbidity, but did not improve survival.122 Pain management is an important priority for patients with advanced PDA. Key sources of pain are from an enlarging primary tumor and/or liver capsular pain associated with metastatic disease. Celiac plexus neurolysis blocks the pain signals transmitted along the celiac plexus infiltrated by primary PDA and has been shown to provide a high level of pain relief in the majority of patients.123 The technique tends to be reserved for patients refractory to opioids or those suffering a high level of toxicity; the benefits of early intervention have never been formally tested.
Weight loss in pancreatic cancer can be due to anorexia, malabsorption, and/or cachexia.124 Palliative interventions include pancreatic enzyme replacement therapy125 and the use of thalidomide to attenuate weight loss in patients with cachexia.126 In addition, PDA is associated with high rates of depression and suicide,127 emphasizing the need to monitor patient distress.
Discussion and future perspectives
Current PDA treatments offer limited benefits; at best, they delay progression and extend survival for very short durations.1,9,128 Among 35 different agents or combinations that have been tested in phase III studies in the last 25 years, only 11% have entered clinical practice.129 Resistance to conventional chemotherapy and radiotherapy is often innate, rather than acquired.9 In addition, the majority of patients diagnosed with PDA are older and frailer compared with patient groups diagnosed with more common cancers. They also tend to have poor functional status associated with late presentation, including biliary and bowel obstruction, cachexia and abdominal pain, thus limiting their tolerance of anticancer therapies. Improving outcomes for patients with PDA is therefore a global urgent unmet need and the focus for both preclinical and clinical research.
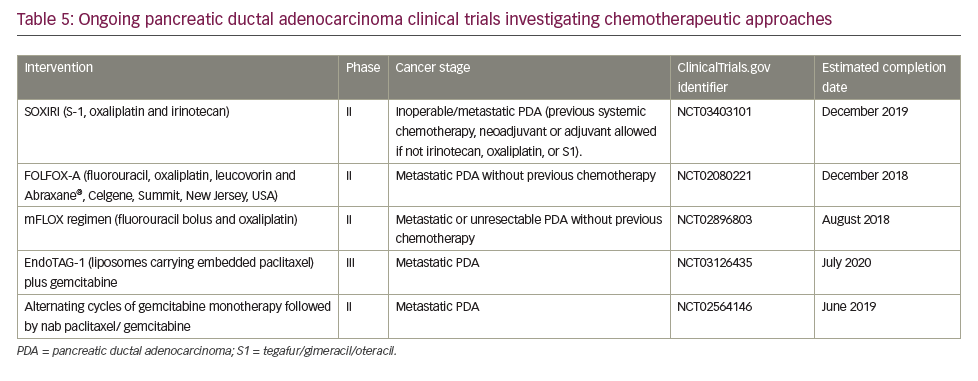
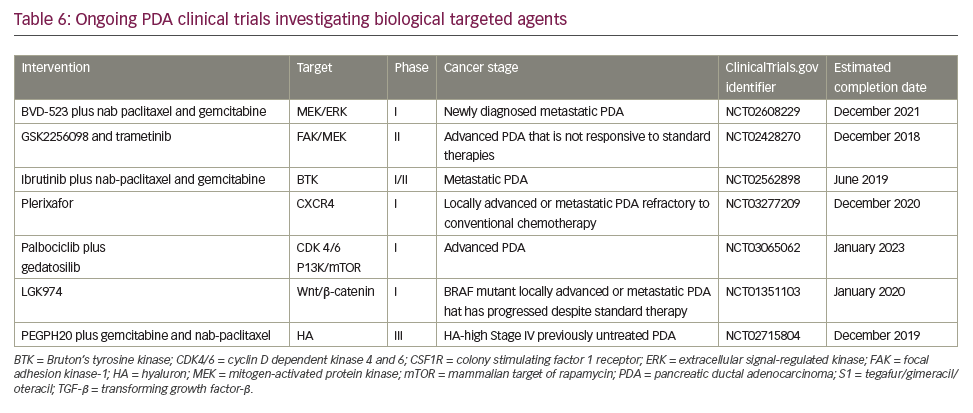
A search of the US National Library of Medicine (ClinicalTrials.gov) in October 2018 identified multiple ongoing trials of chemotherapy agents aiming to adapt doublet (gemcitabine + nab-paclitaxel) and triplet (FOLFIRINOX) therapies to improve outcomes in PDA (Table 5). Biologically targeted agents have been extensively evaluated as a therapeutic strategy in PDA. Despite promising early studies, agents targeting epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), insulin-like growth factor 1 receptor (IGF-1 R), vascular endothelial growth factor (VEGF)/VEGF receptor (VEGFR), NOTCH and WNT have proved ineffective.129 New targets are being evaluated particularly focusing on the microenvironment and mechanisms to amplify T-cell immune response (Table 6).
Immune checkpoint blockade has, to date, proved a disappointing therapeutic strategy for PDA. Neither anti-CTLA4 nor anti-PD1 antibodies have shown activity as single agents overall.130 The only exception so far is the slight activity of pembrolizumab in microsatellite instability-high tumors,119 which represent <5% of all PDA.131,132 One serious challenge for immunotherapy in PDA is that it has a non-inflamed microenvironment with a dearth of T cells and the tumor mutational burden is low relative to other cancers, such as melanoma and lung cancer, against which checkpoint inhibitors are proving most effective. Despite this, numerous clinical trials are currently investigating immunotherapy approaches, most of which are at early stages of development and results are awaited with much interest (Table 4).
An integrated approach using both germline testing and somatic analyses of tumor tissues and next-generation sequencing may assist with the choice of treatment for individual patients in the future.49,133 Some key novel comprehensive national research strategies are currently aiming to develop molecular stratified treatment approaches within clinical trials and in regular clinical practice. These initiatives include Precision Promise in the USA, which is a coordinated and adaptive randomized clinical trial platform that allows multiple novel therapies to be evaluated and aims to expedite their approval for use in PDA.134 In the UK, Precision Panc is establishing a coordinated system aiming to increase the speed of scientific discovery and clinical trials and to develop personalized treatments in order to rapidly improve survival in PDA.135 These systematic approaches to molecular profiling and molecular stratification of PDA involve considerable investment and may well yield significant benefits in the longer term.
Conclusion
PDA has been described as an ‘insidious clinical syndrome’9 that has one of the worst cancer outcomes. Although some environmental and lifestyle factors are implicated in cause, no systematic preventative strategies are in place. Biomarkers to aid detection and treatment are lacking. Most patients present with unresectable disease and despite optimal systemic therapy, life expectancy for most patients remains <1 year. Despite this poor experience, research into PDA molecular pathophysiology is generating increasing understanding of how this cancer grows and evades intervention. In addition, many new targets are emerging which are being exploited as novel therapeutic targets. Clinical trials, some of which have been summarized here, therefore offer our patients the best hope for the future.