Cholangiocarcinoma constitutes approximately 3% of all gastrointestinal malignancies, and the incidence is on the rise, especially the intrahepatic subtype. These tumors are anatomically broadly classified into intrahepatic, perihilar, and distal cholangiocarcinoma. Recent understanding of tumorigenesis pathways has shown that there exists notable differences in molecular pathogenesis between the intrahepatic versus extrahepatic versus gallbladder cancers.1 For instance, a genomic analysis of 489 cholangiocarcinoma tissue specimens revealed notable genetic heterogeneity of the tumors, which signifies the variations in molecular profile depending upon the location and etiology.2 In addition, a comprehensive evaluation of 1,104 cholangiocarcinoma tumor specimens showed that the most common genes (in at least 10% of the specimens analyzed) involved in the tumorigenesis were TP53, CDKN2A/ 2B, KRAS, ARID1A, SMAD4, BAP1, isocitrate dehydrogenase (IDH)1, PBRM, and fibroblast growth factor receptor 2 (FGFR2) (in the order of highest to lowest frequency).3 Interestingly, the study identified potentially targetable aberrations in at least 43% of the tumor samples analyzed, reinforcing the potential role of precision medicine in advanced cholangiocarcinoma.
Initially, molecular therapies targeting epidermal growth factor receptor (EGFR) and vascular endothelial growth factor receptor (VEGFR) pathways did not show encouraging results in unselected populations. For example, although EGFR protein overexpression has been implicated in 11–27% of intrahepatic and 5–9% of extrahepatic cholangiocarcinoma, EGFR inhibitor erlotinib showed only a modest benefit in unselected cohort of cholangiocarcinoma.4 Similarly, cabozantinib, a mesenchymal epithelial transition (MET) inhibitor, also yielded disappointing results in a phase II study involving 19 patients with cholangiocarcinoma.5 In contrast, encouraging results were seen in patients with identified mutational targets, especially in tumors harboring IDH1/2 mutations, FGFR2 fusions in intrahepatic cholangiocarcinoma, and human epidermal growth factor receptor 2 (HER2) mutations in gallbladder cancer.6 These mixed results show that a better knowledge of molecular pathogenesis and advancements in the development of targeted therapy offers hope that we may improve outcomes in a subset of patients with advanced cholangiocarcinoma. Among the newly discovered molecular alterations, targeting IDH1/2 mutations, FGFR2 fusions, RAS-MAPK pathway activation, BRCA1/2 mutations, NTRK fusions, and HER2 receptors, there is great promise for improving the future management of cholangiocarcinoma. In tumors with high microsatellite instabilities or mismatch repair deficiencies irrespective of programmed death ligand 1 (PD-L1) expression, immunotherapy, alone or in combination with targeted agents and chemotherapy are currently being evaluated. This article reviews the key molecular pathological pathways involved in the tumorigenesis of cholangiocarcinoma and focuses on the molecular targeted therapies acting on these pathways.
Molecular pathogenesis and targeted therapies being evaluated in cholangiocarcinoma
Molecular studies have determined the key differences in the tumorigenesis of cholangiocarcinoma are based on anatomical subtype. As such, a comprehensive transcriptome analysis and whole exome sequencing of these tumors have shown distinct genomic aberrations in intrahepatic, perihilar, and distal subtypes.7 A study by Nakamura et al. showed that the aberrations implicated in the tumorigenesis of intrahepatic cholangiocarcinoma involved IDH1, IDH2, FGFR1, FGFR2, FGFR3, EPHA2, and BAP1 genes.7 On the contrary, there was a preferential involvement of AT-rich interactive domain-containing protein 1B (ARID1B), ELF3, polybromo 1 (PBRM1), protein kinase cAMP-activated catalytic subunit alpha (PRKACA), and PRKACB genes in perihilar and distal subtypes.7 This clearly shows that targeted therapies should be tailored to the specific subtype of cholangiocarcinoma, and hence the concept of personalized medicine based on the subtype of cholangiocarcinoma.
Isocitrate dehydrogenase mutations
IDH (isoforms 1, 2, and 3) is involved in the conversion of isocitrate to α-ketoglutarate in Krebs cycle. The key difference between these isoforms is that IDH isoform 1 is limited to cytoplasm whereas isoforms 2 and 3 are located in mitochondrion. Epigenetic alterations of IDH1/2 genes are implicated in about 14% of intrahepatic subtypes, which leads to increased conversion of isocitrate to D-2-hydroxyglutarate. D-2 hydroxyglutarate acts as an oncometabolite, which inhibits α-ketoglutarate dependent dioxygenases, increasing levels of TP53 proteins, leading to hypermethylation of DNA, potentiating tumorigenesis by altering cell differentiation.8,9
Ivosidenib (AG-120) is an oral IDH1 inhibitor which was evaluated in a dose escalation and expansion phase I trial involving 73 patients with cholangiocarcinoma who failed first-line therapy with gemcitabine and cisplatin doublet.10 Ivosidenib resulted in encouraging outcomes in terms of tolerance, overall response rate of 62%—in which 56% had stable disease and the remaining 6% had partial response—which translated to a progression-free survival of 40% in 6 months. At a molecular level, ivosidenib showed morphological alterations in IDH1-mutated cholangiocytes.11 Ivosidenib is currently being evaluated in a phase III multicenter, randomized clinical trial (ClarIDHy trial) in 185 patients with advanced intrahepatic cholangiocarcinoma harboring IDH1 mutations who failed one or two lines of chemotherapy. The trial has a primary endpoint of progression-free survival, while the secondary endpoints include drug tolerance and overall survival (OS; ClinicalTrials.gov identifier: NCT02989857).12 Interim results of the trial were presented at the European Society of Medical Oncology (ESMO) 2019 annual conference.13 The qualified patients were randomized to either ivosidenib 500 mg or placebo. Notably, the patients enrolled on to the placebo arm who had radiological progression during the course of the study were allowed to cross over to ivosidenib arm. Ivosidenib resulted in promising outcomes in terms of progression-free survival (median: 2.7 versus 1.4 months; hazard ratio [HR] = 0.37; 95% confidence interval [CI] 0.25–0.54; p<0.001)—32% and 22% of the ivosidenib cohort had progression-free survival at the end of 6 and 12 months, respectively.
These encouraging results translated to a disease control rate of 53%, in which 51% had stable disease and 2% had partial response. This is in contrast to none of the patients in the placebo arm having progression-free survival at the end of 6 months, while no patients were left in placebo group at the end of 12 months. Ivosidenib showed a trend towards improvement in OS as compared to that of placebo (10.8 versus 9.7 months). On exploratory analysis using the Rank-Preserving Structural Failure Time method as if the patients were never crossed over, statistically and clinically meaningful differences were seen in estimated OS (10.8 versus 6.0 months, p<0.001). Ivosidenib was well-tolerated with no clinically meaningful differences in grade III side effects between the two arms (46% versus 36%). The phase III trial is the first of its kind that has demonstrated promising results with targeted therapy in patients with cholangiocarcinoma, which has opened doors to precision medicine in this dismal cancer.
Fibroblast growth factor receptor 2 genetic fusions
FGFR2 fusions with other genes such as BICC1, PPHLN1, MGEA5, transforming acidic coiled-coil containing protein 3 (TACC3), and coiled-coil domain-containing protein 6 (CCDC6) are implicated in the pathogenesis of 13–20% of intrahepatic cholangiocarcinoma tumors.14,15 Mutations in FGFR1 and FGFR3 have also been encountered in the carcinogenesis of intrahepatic subtype.7 In addition, upregulation of FGFR 1, 2, and 4 were demonstrated in the cholangiocarcinoma cell lines, which was thought to be mediated by a transcriptional cofactor, yes-activated protein.16 These fusions are thought to activate the downstream signaling pathways of RAS-MAPK and JAK-STAT.17–19 Furthermore, FGF ligand has been implicated in the MEK1/2 phosphorylation contributing to migration of cholangiocarcinoma cells.20 Some FGFR2 fusions such as FGFR2–AHCYL1, and FGFR2–KIAA158 were associated with KRAS mutations.17,21 In addition, BAP1, TP53, and CDKN2A/B mutations were the most common co-existing mutations. While co-existing BAP1 mutations do not influence median OS, co-existing mutations with TP53 and CDKN2A/2B are associated with inferior median OS (p=0.04).22 Notably, FGFR2 fusions were shown to be associated with female sex (13% versus 4%), younger age at diagnosis (52 versus 65 years) and better median OS (123 versus 37 months).15
FGFRs are known to harbor tyrosine kinase domains, as such, tyrosine kinase inhibitors targeting FGFRs have been extensively evaluated in intrahepatic cholangiocarcinoma.23 In a phase II multicenter clinical trial, infigratinib (BGJ398) showed promising outcomes in 61 patients with advanced cholangiocarcinoma that failed first-line gemcitabine-based chemotherapy.24 Among these 61 patients enrolled, 48 patients were shown to harbor FGFR2 fusions, eight and three patients had mutations and amplifications of FGFR2, respectively. The primary endpoint of the study was to evaluate the overall response rate, which was accounted to be 14.8% in the entire cohort, while the patients harboring FGFR2 fusions had an overall response rate of 18.8%. Infigratinib resulted in a disease-control rate of 75.4% which translated to a median progression-free survival of 5.8 months. While the drug was well tolerated, the most common side effects noted were hyperphosphatemia, fatigue, stomatitis, and alopecia primarily resulting from blocking other FGFR pathways.
The authors presented (at the ESMO 2018 annual meeting) updated data of the same trial that enrolled 71 patients with advanced intrahepatic cholangiocarcinoma and with FGFR2 fusions, which showed partial response, stable disease, and progressive disease in 24%, 58%, and 11%, respectively.25 One has to be cautiously optimistic with these FGFR-targeting tyrosine kinase inhibitors given the development of resistance mechanisms, which may be attributed to the drug-binding site kinase mutations (pN549H, pL617V, pE565A, pK641R, pN549K, pV564F, and pK659M) and downstream pathway, PTEN/PI3K alterations. Cellular studies have shown that ponatinib and dovitinib, non-selective tyrosine kinase inhibitors, have promising activity at these point-mutations (pN540 and p549K).26
Derazantinib (ARQ-087) is another oral pan-FGFR tyrosine kinase inhibitor that has been evaluated in cholangiocarcinoma in phase I and II trials. In a phase I/II trial involving 29 patients with cholangiocarcinoma, tumor burden reduction of 10–29% was seen in three out of 12 patients with FGFR2 fusions.27 Among the 12 patients with FGFR2 fusions, six patients had a disease control for >4 months. The study had an expansion phase that enrolled a total of 29 patients with advanced cholangiocarcinoma that harbored FGFR2 fusions and objective response rate of 21%.28 A single-arm phase III trial is currently enrolling patients with FGFR2 fusion-positive advanced cholangiocarcinoma that failed one or more lines of systemic therapy agents (FIGURE trial; ClinicalTrials.gov identifier: NCT03230318).
Futibatinib (TAS-120) is a covalent and irreversible inhibitor of FGFRs currently being evaluated in advanced cholangiocarcinoma. The drug was evaluated in 19 patients with cholangiocarcinoma (17 of intrahepatic subtype and two were of extrahepatic) harboring FGF/FGFR aberrations and half of the cohort had partial response. In addition, among three patients with FGFR2 fusions who received a prior therapy with another FGFR inhibitor, one patient was seen to have partial response and another stable disease.29 Most recent updates on the same study demonstrated a tumor shrinkage in 71%, with an objective response of 25% in tumors harboring FGFR2 fusions.30 Given the irreversible binding nature of the drug to FGFR, it is currently being evaluated in the patients who had progressive disease with other FGFR inhibitors (ClinicalTrials.gov identifier: NCT02052778).
Pemigatinib is a potent FGFR inhibitor with a selective activity against FGFR 1, 2, and 3 kinases. Pemigatinib showed a promising overall response rate in patients with advanced cholangiocarcinoma harboring FGFR2 fusions or rearrangements. Recent data from the FIGHT 202 trial showed that pemigatinib resulted in an overall response rate of 35.5%, with a disease control rate of 80% in 107 patients with advanced cholangiocarcinoma harboring FGFR2 fusions or rearrangements.31 On the contrary, no clinically meaningful responses were seen in other patients with FGFR gene alterations or other genetic alterations. Given these encouraging results, the United States Food and Drug Administration accepted the new drug application of pemigatinib in FGFR2 fusion/rearrangement harboring cholangiocarcinoma for a priority review.
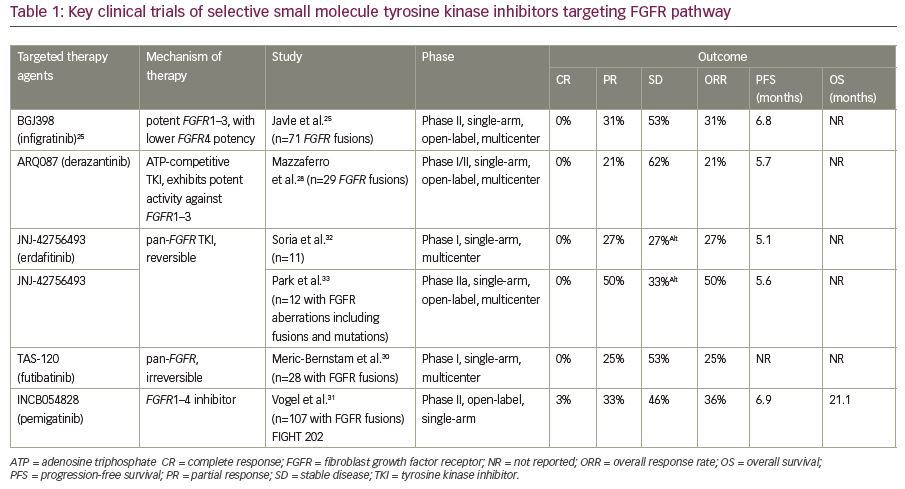
Table 1 summarizes key clinical trials evaluating FGFR inhibitors—infigratinib, derazantinib, erdafitinib, and futibatinib—in patients with advanced cholangiocarcinoma.25,28,30–33 Other tyrosine kinase inhibitors being evaluated in clinical trials include, but are not limited to, ponatinib (ClinicalTrials.gov identifier: NCT02265341, NCT02272998), erdafitinib (ClinicalTrials.gov identifier: NCT02699606), E7090 (FGFR 1, 2 and 3 receptor inhibitor), and rogaratinib (ClinicalTrials.gov identifier: NCT01976741).21,34
Multi-kinase tyrosine inhibitors blocking HGF/c-MET signaling, EGFR, MAPK pathways
MET overexpression is implicated in the tumorigenesis of the intrahepatic subtype and perihilar/distal subtypes due to its role in angiogenesis and cell migration.35,36 In addition, c-MET is known to activate the downstream signaling pathways—JAK-STAT, AKT, and MAPK pathways.37 Similarly, EGFR has also been implicated in cell migration and angiogenesis.15 EGFR overexpression has been implicated in the carcinogenesis of cholangiocarcinoma with the highest level of expression observed in the intrahepatic subtype (38–100%).31 Aberrant phosphorylation and overexpression of EGFR activates MAPK/ERK and p38 pathways triggering the tumor growth and inhibition of apoptosis, thereby promoting the tumorigenesis.32 In intrahepatic cholangiocarcinoma, MET amplification was shown to be associated with short OS and >5 cm tumor size.38
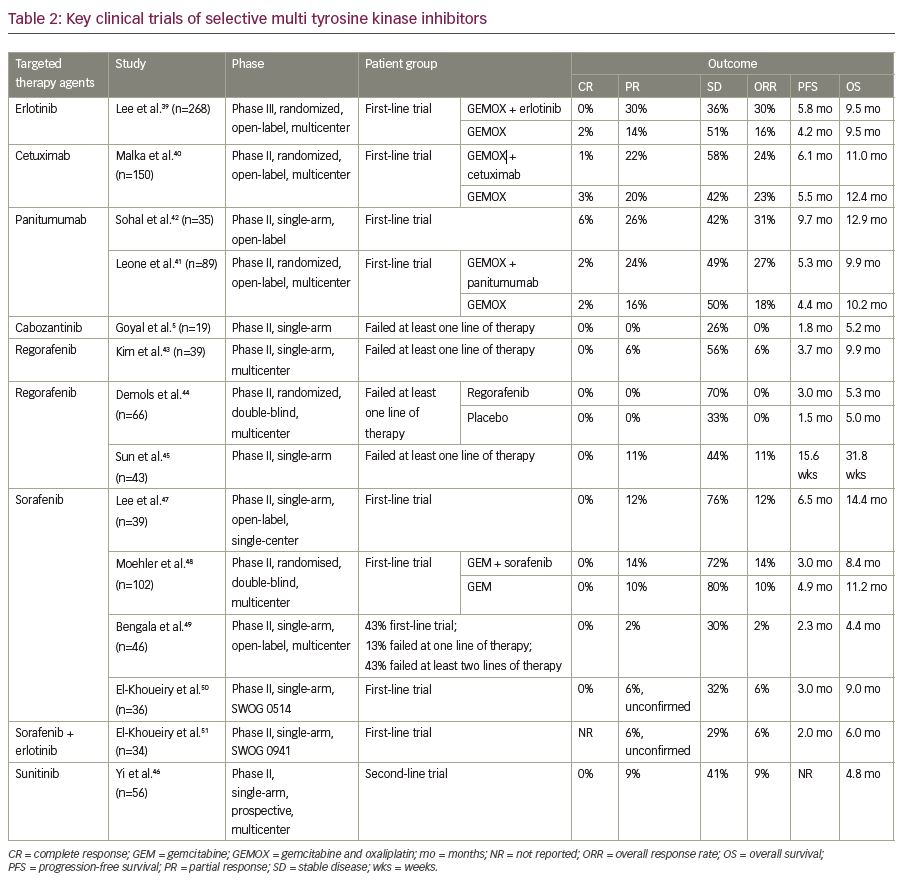
Erlotinib, a tyrosine kinase inhibitor targeting the EGFR pathway was evaluated in 268 patients with advanced cholangiocarcinoma in combination with gemcitabine and oxaliplatin doublet in a phase III trial. Though there has been an objective response in a greater percentage of patients who received erlotinib, the objective response did not translate to progression-free survival benefit.39 Similar unsatisfactory results in terms of progression-free and median OS were seen with the addition of EGFR antibody, cetuximab.40 On the contrary, addition of panitumumab, another monoclonal antibody targeting EGFR, to the combination regimen of irinotecan and gemcitabine, resulted in 74% disease control rate. Moreover, the addition of these agents to chemotherapy in the presence of wild-type KRAS mutations was demonstrated to be not so effective in terms of efficacy.41 While anti-EGFR therapy with panitumumab and cetuximab yielded mixed results,40–42 the use of the c-MET inhibitor cabozantinib showed limited activity at the expense of considerable toxicity in non-biomarker driven studies.5 Hence, given the inconsistent results with EGFR and c-MET inhibitors, the role of these agents is yet to be evaluated in larger cohort. Regorafenib, a multi-kinase inhibitor was evaluated in phase I/II trials in patients with advanced cholangiocarcinoma with modest results in terms of overall and progression-free survival (Table 2).5,39–51 Sunitinib demonstrated a progression-free survival benefit of 1.7 months in a single-arm phase II trial,46 whereas sorafenib did not yield encouraging results in various studies as detailed in Table 2.47–51
On the contrary, a combination of BRAF inhibitor, dabrafenib and MEK inhibitor, trametinib has shown encouraging efficacy results in 33 patients with BRAFV600E mutation-positive advanced cholangiocarcinoma.52 This combination regimen resulted in a duration of response >6 months in 54% of the study cohort, translating to a median OS of 11.2 months. These encouraging results opened doors to the possible role of BRAF inhibitors in BRAFV600E mutation-positive advanced cholangiocarcinoma.
HER2/neu (ERBB2) and HER3 receptor tyrosine kinase pathway signaling
HER2 overexpression has been implicated in the pathogenesis of 10–16%, 5–9%, and 1% of gallbladder cancers, extrahepatic, and intrahepatic cholangiocarcinoma, respectively.14 HER2 amplifications trigger the HER2/neu receptors thereby activating the downstream signaling pathways RAS-RAF-MEK-ERK or PI3k-AKT-mTOR resulting in the proliferation of tumor cells. Despite the presence of HER2 amplifications in a small percentage of patients with advanced cholangiocarcinoma, preliminary data in the form of a case series are encouraging.53 This case series evaluated the role of HER2-targeted therapy with trastuzumab, lapatinib, and pertuzumab in five patients with advanced cholangiocarcinoma and nine with gallbladder cancer. Though the patients with advanced cholangiocarcinoma did not show any radiological response, one, four, and three patients with gallbladder cancer had a complete response, partial response, and stable disease, respectively. Mixed responses were seen with lapatinib in a patient who harbored Val777Leu mutation in the HER2 kinase domain. While the preliminary data on HER2-targeted therapy looks promising, more detailed information and/or more robust data through clinical trials is much awaited (ClinicalTrials.gov identifiers: NCT00101036, NCT02836847).
Immunotherapy
Immunotherapy with programmed cell death-1 (PD-1), PD-L1, and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibitors has emerged as a promising therapeutic option in various cancers such as melanoma, non-small cell carcinoma, and renal cell carcinoma, to name a few. In a comprehensive genomic analysis of 3,364 cholangiocarcinoma tumor specimens, 3.5% of the tumors showed >10 mutations/megabase (mb), whereas 1.4% showed >20 mutations/mb.54 In this analysis, PD-L1 expression was seen in 9% of the samples analyzed, substantiating the possible role of checkpoint inhibitors. Patients with advanced cholangiocarcinoma with mismatch repair deficiency, high micro-satellite instabilities, with or without PD-L1 expression should be encouraged to participate in clinical trials evaluating the role of these agents.
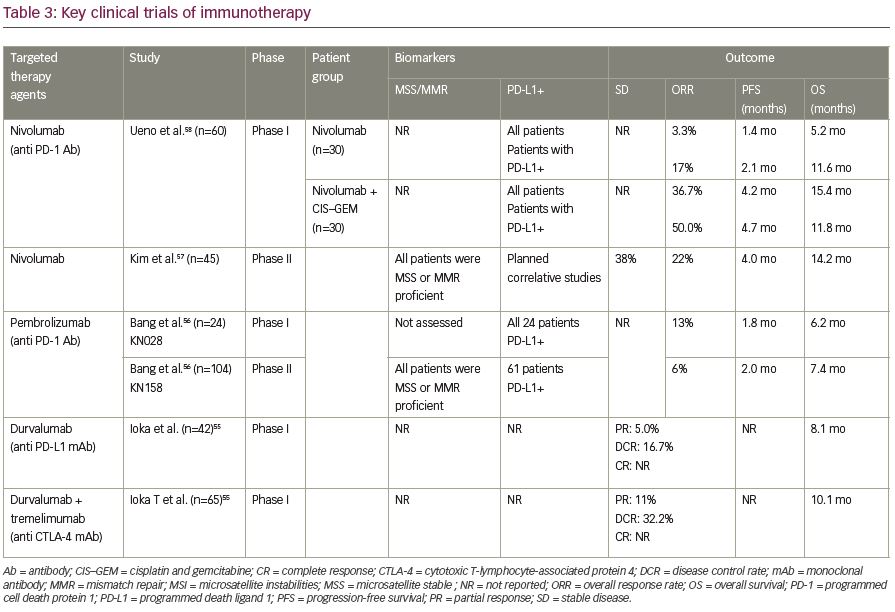
Immune checkpoint inhibitors including nivolumab, pembrolizumab, and durvalumab (alone or in combination with tremelimumab) were evaluated in phase I/II trials in patients with advanced cholangiocarcinoma.55–58 The preliminary data showed the role of checkpoint inhibitors in advanced cholangiocarcinoma. For instance, nivolumab resulted in 6- and 12-month OS rate of 71.4% and 52.3%, respectively in 45 patients with advanced cholangiocarcinoma that progressed on at least one-line of systemic chemotherapy (44% failed two lines).57 Furthermore, response rates were up to 22% with promising OS of 14 months in refractory in patients with microsatellite stable advanced bile duct tumors. However, the results could not be duplicated with other single-agent checkpoint inhibitor studies which are outlined in Table 3.55-58 The response rate for other checkpoint inhibitors were around 10%, irrespective of PD-L1 positivity.56
However, nivolumab in combination with systemic chemotherapy of gemcitabine and cisplatin in a Japanese cohort of 30 patients with advanced cholangiocarcinoma, seemed to demonstrate promising results.58 While nivolumab monotherapy resulted in a modest response in terms of objective response, the combination of nivolumab with gemcitabine and cisplatin resulted in a median OS of 15.4 months and objective response of 37% (11 out of 30 patients). Table 3 summarizes key clinical studies with immunotherapy agents in advanced cholangiocarcinoma. The major challenge is finding a better biomarker that can predict a promising response to immunotherapy.
Other potential druggable pathways and targeted therapies in advanced cholangiocarcinoma
In addition to the above detailed genetic aberrations and pathways, BRCA1/2 mutations have been implicated in a small percentage of cholangiocarcinoma tumors.59 A molecular profiling of 1,288 tumor specimens of cholangiocarcinoma showed that 3% of the specimens had BRCA2 mutations while 0.6% had BRCA1 mutations. Poly eno (ADP-ribose) polymerase (PARP) inhibitors have shown promising results in other BRCA-mutated malignancies such as ovarian and breast cancers.60 Notably, on a retrospective analysis of 18 patients with BRCA-associated advanced cholangiocarcinoma, PARP inhibitors showed promising efficacious outcomes as compared to standard first-line platinum-based chemotherapy.61 A couple of clinical trials (ClinicalTrials.gov identifiers: NCT04042831, NCT03639935) are currently enrolling patients with BRCA-associated advanced cholangiocarcinoma to study the efficacy and safety of PARP inhibitors in this specific subgroup.
NTRK fusions have also been implicated in the pathogenesis of cholangiocarcinoma via activation of downstream MAPK signaling pathway.62 NTRK inhibitors, entrectinib and larotrectinib, are approved by the United States Food and Drug Administration for the use in patients with tumors harboring NTRK fusions who have failed standard first- and second-line therapies. These inhibitors are currently being evaluated in advanced cholangiocarcinoma clinical trials (ClinicalTrials.gov identifiers: NCT03212274, NCT02576431).
Conclusion
As our knowledge around the tumorigenesis of cholangiocarcinoma progressed and we welcomed the advent of personalized medicine in the field of oncology, many targeted therapies have begun to be evaluated in this rare but dismal cancer. As detailed above, a considerable number of studies have shown encouraging results. Agents targeting FGFR2 fusions, IDH1/2 mutations, HER2 amplifications, BRAF mutations, and NTRK fusions have been particularly encouraging. The promising efficacy data from the ClarIDHy trial clearly showed that precision and personalized medicine is indeed applicable to advanced cholangiocarcinoma.12 Given these encouraging results, it is reasonable to recommend mutation testing for FGFR2 fusions, IDH1/2 mutations, HER2 amplifications, BRAF mutations, and NTRK fusions in patients and they suggest that we should encourage participation in respective clinical trials. For optimal outcomes, clinical trials should be tailored to tumor biomarkers and mutations. We conclude, with optimism, that we will reach the point where we use personalized medicine as a standard of care in advanced cholangiocarcinoma. To make this dream come true we need extensive collaborative efforts to enroll patients in global clinical trials.