H-Ras, K-Ras, and N-Ras are the main members of Ras superfamily, which bind small molecules GTP and GDP interchangeably and can hydrolyze GTP to GDP. The Ras superfamily consists of more than 150 proteins and these can be classified under at least five sub-families viz: the Ras, Rab, Rho, Ran, and Arf families.1 Ras oncoproteins act as typical molecular switch by alternately binding to GTP and GDP molecule and has intrinsic GTPase activity. It remains in an active state when bound to GTP and switches to an inactive state by binding to GDP and thus controls the expression of the downstream genes. Ras signaling is an important intracellular signaling pathway that plays a role in cellular proliferation and differentiation, survival, and gene expression.2–4 Ras oncoprotein has also been implicated in the development of cancer by either having increased intensity or prolonged signaling mechanism.5 This may happen either due to a mutation in the Ras-GTPase domain, which renders it constitutively inactive (GDP-bound state), or due to activating mutation in growth factor receptors that act upstream of Ras or due to aberrant RAS effector activation. Due to the elevated level of Ras signaling in tumor growth and progression, Ras proteins and its downstream effector proteins may serve as promising therapeutic targets against cancer. In this review article we shed light on normal Ras signaling as well as its aberrant signaling in tumors and proposed various strategies for its inhibition.
Normal Ras Signaling Pathways
The three human RAS genes encode four proteins with a size of ~21 kDa: H-Ras, N-Ras and the splice variants K-Ras4A and K-Ras4B. A newly synthesized Ras protein is a soluble cytoplasmic protein, which needs to undergo post-translation modifications to associate with particular lipid membrane. These modifications occur at the carboxyl terminal ‘CAAX’ box (denoting amino acid sequence cysteine-aliphatic-aliphatic-X residue). The first step involved is the attachment of 15 carbon farnesyl to the cysteine residue of the ‘CAAX’ box.6 Next, an endopeptidase Rce1 cleaves off three terminal amino acid residues and the resulting isoprenylated cysteine residue is methylated by the isoprenylcysteine carboxyl methyltransferase (ICMT) as shown in Figure 1.7 H-Ras and K-Ras undergo additional palmitoylation modification.8 These post-translational modifications are necessary for binding to lipid membrane to execute their biologic functions.
The three-dimensional structures of Ras proteins with bound GTP and GDP and their mutant variants were determined through X-ray crystallography in 1990.9–11 The Ras protein consists of a hydrophobic core of six β sheets and five α helices that are interconnected by a series of 10 loops. Five of these loops determine the high-affinity nucleotide interactions of Ras and regulates GTPase activity. The GTP γphosphate is stabilized by interactions that are established with the residues Lys16, Tyr32, Thr35, Gly60, and
![]()
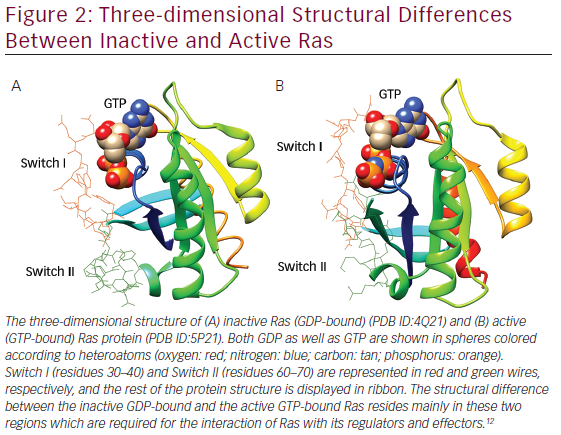
Gln61 of loops. Gln61 is a key residue that stabilizes the transition state of GTP hydrolysis to GDP, in addition to participating in the orientation of the nucleophilic attack that is necessary for this reaction. It has been seen that oncogenic mutations of Gln61 reduce the intrinsic GTP hydrolysis rate, thereby rendering the Ras protein constitutively active. The structural differences in GDP-bound Ras (inactive state) and GTP-bound Ras (active state) lies mainly in highly dynamic regions termed Switch I (residues 30–40) and Switch II (residues 60–70) as shown in Figure 2, which are required for the interaction of Ras with both upstream as well as downstream partners. The binding of GTP brings change in structural conformation of side chain of Switch I, via inward reorientation of side chain of Thr35, which facilitates its interaction with the GTP-γ phosphate as well as the Mg2+ ion. Similarly, the γ-phosphate induces significant changes in the conformation of Switch II through the interaction it establishes with Gly60.12
Besides its normal function, such as cellular proliferation and differentiation, survival, and gene expression, gain-of function mutations of H-, N-, K-Ras have been found in different types of human cancers.12,13 Besides, aberrant Ras signaling is also implicated in several developmental disorders, known as the cardio-facio-cutaneous diseases (i.e., neurofibromatosis-type I [NF-1], Costello syndrome, and Noonan syndrome).14
The normal biological functioning of RAS proteins as described before require post-translational modification, guided by many important enzymes described earlier. These RAS preprocessing enzymes can serve as attractive drug targets. The activity of RAS is largely determined by the type of cofactors that it binds. When it is bound to GTP, it is active and can recruit downstream target proteins, but when it binds GDP, it is rendered inactive and fails to interact with the downstream effectors. This association of RAS with GTP or GDP is mediated by two enzymes: guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs). GEFSs catalyze the exchange of GDP for GTP whereas GAPs increase the rate of GTP hydrolysis to GDP plus phosphate.15
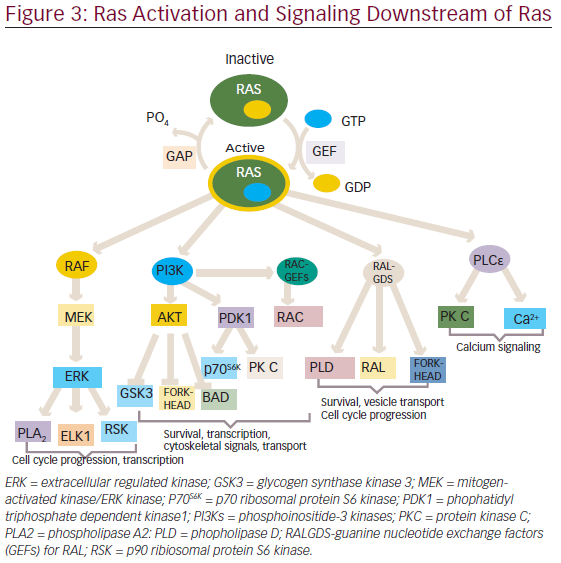
The activated RAS protein can bind and activate downstream effectors, which ultimately produce appropriate signals, such as cell proliferation, survival, and other physiological functions through the activation of various pathways (see Figure 3).
One of the first mammalian effector of RAS, which has been well studied and characterized is the protein serine/threonine kinase, is RAF.16,17 There are three closely related RAF proteins, CRAF1, BRAF, and ARAF, which are known to be activated by RAS-bound GTP.18 The activated RAF can phosphorylate and activate the downstream targets, such as mitogenactivated protein kinases 1 and 2 (MEK1 and MEK2).19 MEK1 and MEK2 can then phosphorylate and activate mitogen-activated protein kinases (MAPKs), such as extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2). The substrates for ERK1/2 include both nuclear as well as cytosolic proteins, of which transcription factors have been widely studied.20,21 ERK phosphorylate (ELK1) belongs to ETS family transcription factors, which, in turn, regulates the expression of FOS.22 Additionally, ERK can also phosphorylate c-JUN.23 The activation of all these transcription factors eventually promotes the cell cycle progression.24
In addition to the RAF/MAPK effector pathway, RAS can also interact with another well-characterized effectors ie., phosphatidyl inositol 3-kinases (PI3Ks).25,26 The activated PI3Ks phosphorylate phosphatidylinositol 4,5-bisphosphosphate (PtdIns(4,5)P2) produce a second messenger phosphatidylinositol-3,4,5-triphosphate(PtdIns(3,4,5)P3), which interacts with several other proteins through pleckstrin homology and other domains.27 Thus, PI3K exercises its control on a large number of downstream target proteins. PI3K regulates the activity of two important kinases PDK1 (3-phosphoinositide-dependent protein kinase-1) and AKT.28,29 PDK1 is important for the activation of many protein kinases of the AGC family, comprising AKT/PKB, p70S6K, some PKCs and RSKs.30–33 AKT has an antiapoptotic function and have been found to be important for survival signals generated by RAS.34,35 Besides, PI3K activation also leads to the activation of RAC, a RHO family protein that not only regulates the actin cytoskeleton but also transcription factors, such as nuclear factor-kappa B( NF-κB).36–38 RAC activation also seems to be important in RAS-induced transformation.39,40
The third well-known effector of RAS include three exchange factors for RAS-related RAL proteins: RAL guanine nucleotide dissociation stimulator (RALGDS), RGL2/RLF, RALGDS-like gene (RGL/RSB2).41–43 With the help of these proteins RAS is able to activate RAL, which in turn activates phospholipase D1, CDC42/RAC-GAP-RAL binding protein1 (RALBP1).44,45 These RALGDS pathway together with AKT contribute to the inhibition of FORKHEAD transcription factors.46 These have been involved in arresting cell cycle progression through activation of cyclin dependent kinase inhibitor, KIPI (also known as p27), and apoptosis through expression of the BIM and FAS ligands.47,48
Phospholipase Cε is another effector of Ras.49 It hydrolysis phosphatidylinositol 4,5-bisphosphosphate to diacylglycerol and inositol-1,4,5-triphosphate, which leads to activation of PKC and calcium mobilization.50
Through the concerted efforts of RAS and its effectors, it is able to regulate a wide array of functions, such as cellular proliferation, survival, apoptosis, and other important physiological processes.
Abnormal RAS Signaling in Tumors
Activating Mutation in RAS
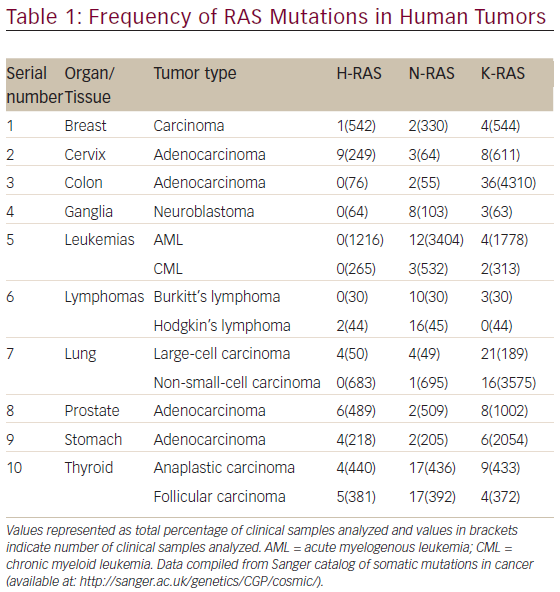
The aberrant RAS signaling in tumors can be contributed by several different mutations, mostly activating mutation in tumor cells: 85 % in K-RAS, 15 % in N-RAS, and <1 % in H-RAS. Table 1 shows the frequency of RAS mutations in various human tumors. It is clear from Table 1 that significantly high rates of Ras mutation (>10 %) was found in tumors, such as colon (adenocarcinoma), leukemias (AML), lymphomas (Hodgkin’s lymphoma), lung (large-cell carcinoma and non-small-cell carcinoma) and thyroid (anaplastic and folllicular carcinoma). It is also evident from Table 1 that the oncogenic mutation predominately affects the K-Ras locus, with oncogenic K-Ras mutations being detected ranging from 16 to 36 % in various human tumors screened. The activating mutations mostly affect the GTPase activity of RAS leading to accumulation of RAS-bound GTP.51,52 These GTP-bound RAS can hyperactivate other downstream effector proteins previously discussed before leading to constitutive abnormal signaling and anarchy within the tumor cell. The impaired ability of Ras mutants to hydrolyze GTP, either intrinsically or in response to GAPs, is responsible for the oncogenic nature of mutations at residues G12, G13, and Q61 in the active site.53
GAP Deletion
Ras remains activated due to loss of GAP-accelerated GTP hydrolysis. One such typical example of GAP mutation is the GAPs, neurofibromin encoded by the NF1 tumor suppressor gene.54 Patients with neurofibromatosis type I inherit onlyone functional NF1 gene and then predisposed to cancer through complete loss of NF1.
Growth Factor Receptor Activation
Ras signaling has also been known to be activated in tumors in which growth factor receptor tyrosine kinase has been overexpressed. The most common example are epidermal growth factor receptor (EGFR) and receptor tyrosine-protein kinase erbB-2 (ERBB2) which are activated and overexpressed in many types of cancer including breast, ovarian, and stomach carcinomas.55
Mutation or Amplification of Ras Effectors It has been found that BRAF is commonly activated by mutation in human tumors, such as melanomas and colon carcinoma. A study of 923 cancer samples reveal that missense mutations occur commonly in the BRAF
gene in approximately 70 % of human malignant melanoma and 15 % of colorectal cancers.56 The major mutation found in tumors is V559E, a substitution of valine by glutamic acid, which occurs in the kinase activation domain resulting in activation of BRAF.56
The PI3K pathway is known to be activated due to amplification or mutation of PIK3CA encoding catalytic p110α subunit of PI3K in ovarian and cervical tumors.57,58 These mutations commonly occur in two conserved regions of the gene, which encode the kinase and helical
domains of the protein. These ‘hot spot’ mutations, H1047R, E545K, and E542K, are nonsynonymous missense mutations that confer constitutive kinase activity.59,60 Second mechanism of PI3K activation occurs by amplication of its downstream target AKT2 in ovarian and breast tumors.61 A somatic missense mutation in the pleckstrin homology (PH) domain of AKT1 (E17K) had been identified in breast, colorectal, and ovarian cancers.62 Additionally, PI3K can be directly activated due to loss of tumor suppressor gene PTEN (phosphatase and tensin homolog deleted on chromosome ten). This gene encodes a lipid phospholipase that dephosphorylate phosphatidyl-3,4,5-triphosphate at position 3 of the inositol ring, which reverses the accumulation of these second messengers caused by PI3K and thus negatively regulates PI3K activity. PTEN is one of the most frequently mutated genes in human cancers.63,64
Ras Inhibition Strategies
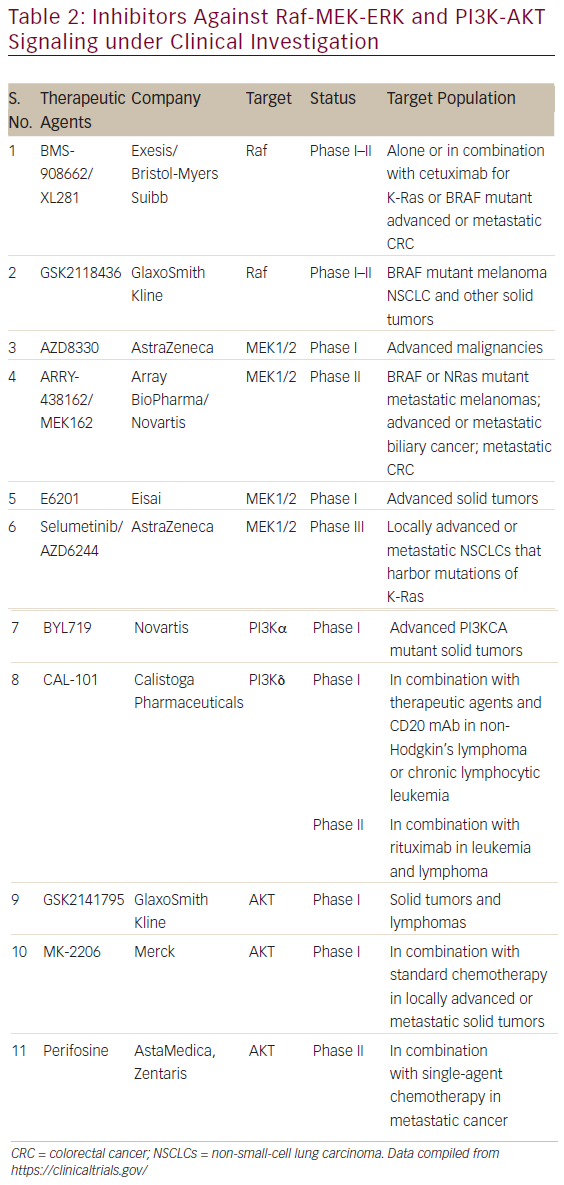
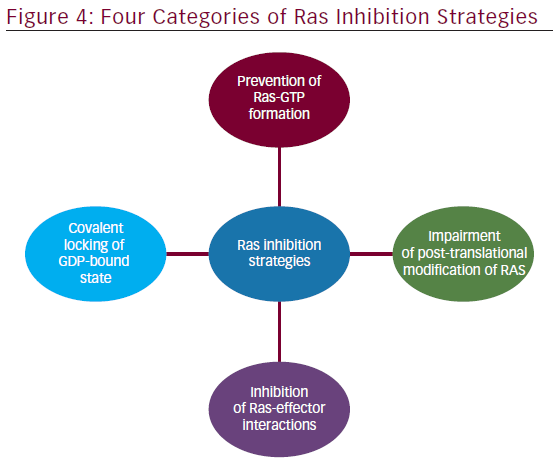
The involvement of aberrant Ras signaling in different kinds of tumors in humans necessitates the development of therapeutic agents, which can restore a normal function in tumor cell. The developments of therapeutics against it have already started and some have successfully managed to enter into phase I–II clinical trials (see Table 2). Targetting the aberrant Ras signaling has largely come from indirect inhibition of Ras effectors, such as AKT, MEK-1/2, PI3K, Raf, etc. This Ras signaling inhibition strategies can be categorized into following four inhibition strategies (Figure 4); each one is discussed briefly below.
Prevention of Ras-GTP Formation
Early studies explored the potency of ATP-competitive kinase inhibitors, which were expected to be used for inhibiting nucleotide binding to Ras. Although the affinity of kinases for ATP are usually in the micromolar range,65 picomolar nucleotide affinities of Ras combined with millimolar intracellular nucleotide pools complicate the use of GTP-competitive inhibitors.66 Consequently, strategies that prevent the initial formation of the Ras–GTP complex were investigated.
Libraries of GTP analogs with alternations at the ribose or nucleotide moiety66 and of pyrazolo[3,4-b]quinoline ribosides67 yielded molecules with moderately increased affinity compared with GDP and relatively weak inhibitory potency.
Covalent Locking of the GDP-bound State
Because of the high affinity of Ras to GDP, nucleotide exchange requires assistance by GEF proteins such as Son of Sevenless (SOS) or the closely related RasGRF1 to facilitate GTP loading. Early examples of GEF inhibitors were identified from a compound library whose members were originally designed to compete with GDP for nucleotide binding. SCH53239 and its derivatives target a hydrophobic pocket close to the nucleotide binding site and inhibit the intrinsic nucleotide exchange.68,69 On the basis of these studies, a series of derivatives with improved potency and water solubility was designed that is capable of inhibiting the RasGRF1-catalyzed nucleotide exchange in vitro (half-maximum in hibitory concentration [IC50] = 35−320 μM).70,71 The orthosteric Ras-GEF interaction inhibitors described above do not discriminate between mutated and wild-type Ras. In an approach to overcome this limitation for mutated K-RasG12C, the thiol function of cysteine 12 was used to covalently trap inhibitors. A set of GDP-derived inhibitors was developed to directly target the nucleotide binding site.72 The most active compound, SML-8-73-1, covalently binds K-RasG12C even in the presence of millimolar concentrations of GDP and GTP. SML-10- 70-1 shows antiproliferative activity in Ras-dependent cells expressing K-RasG12C (EC50 = 27−47 μM).73
Ostrem et al.74 reported covalent inhibitors that are selective for mutant cysteine over the wild type as it relies on trapping of the thiol group in Cys12 in common oncogenic mutant (K-Ras G12C) using disulphide-l fragment-based screening approach. Binding of these inhibitors to K-Ras (G12C) destabilize the native nucleotide preference to favor GDP over GTP and impedes binding to Raf.
Inhibition of Ras-effector Interactions
Protein–protein interfaces (PPIs) between Ras-GTP and effectors initiate various downstream signaling cascades. Remarkably, Ras–GTP exists in at least two distinct conformational states, which interconvert with rate constants on the millisecond timescale.75 Whereas state 2 represents a conformation with high affinity for effector binding, the affinities for effectors exhibited in state 1 are reduced.76,77 However, state 1 exhibits surface cavities potentially accessible to small molecules that could stabilize this conformation and thereby inhibit interactions with effector proteins. Zn2+-cyclen, Cu2+-cyclen, and bis(2-picolyl)amine complexes bind H-Ras–‘GTP’ with millimolar affinity and stabilize the ‘noneffector binding’ state 1.78,79 The feasibility of orthosteric Ras-effector interaction inhibition was shown with antibody fragments that block effector interaction sites of H-Ras–GTP.80 In this setup, disruption of mutant Ras-effector interactions is sufficient to prevent tumor initiation in a transgenic mouse model of lung cancer.81 Small peptide–based inhibitors lacking drug-like properties have been used to disrupt Raseffector interactions.82,83
Impairment of Post-translational Modification of Ras
Ras-dependent signaling requires the correct intracellular localization of Ras proteins predominantly at the plasma membrane, mediated by membrane-anchoring lipid residues at the C terminus. Therefore, impairment of Ras localization has been explored to inhibit oncogenic Ras signaling.84 Ras proteins are equipped with lipid groups through a series of post-translational modifications, which include cysteine S-farnesylation, proteolysis, and carboxymethylation at the C terminus of all Ras isoforms and additional cysteine S-palmitoylation of H-Ras and N-Ras.85
FarnesyltransfeRase inhibitors (FTIs) interrupt this biosynthetic sequence, leading to nonlipidated cytosolic Ras. Several FTIs reached late-stage clinical trials but ultimately failed, mostly because of alternative geranylgeranylation of the K-Ras and N-Ras isoforms.86 Treatment with
geranylgeranyl transfeRase inhibitors87 or dual prenylation inhibitors88 did not show clinical efficacy. However, a number of studies report promising preclinical and clinical results for FTIs as single agents or in combination with other conventional anticancer agents.89 FTIs may also show positive clinical responses in the treatment of H-Ras-dependent cancers and tumors that rely on other farnesylated proteins for survival.87
Conclusion
Ras signaling pathways constitute central drivers of cancer development and therefore strategies have been sought for development of potent Ras inhibitors effective in vivo as well. Some inhibitors against Ras effectors such as Raf, MEK1/2, PI3K, AKT, etc. have already entered into clinical trials, which suggests the relevance of Ras signaling pathways in cancer therapeutics. Since Ras signaling pathway is a complex network controlled by several feedback loops, blocking a single pathway may not be adequate to achieve a reduction of Ras signaling to a therapeutically significant level. Therefore, combined therapies targeting Ras and other oncoproteins in parallel may be required to control tumors.