Adult-type diffuse low-glade gliomas (LGGs) (World Health Organization [WHO] grade 2) represent 5–10% of all primary brain tumours.1 The median age of onset is in the third and fourth decades of life. Common clinical presentations include seizures, focal neurological deficits, and new-onset headaches.2 Magnetic resonance imaging (MRI) demonstrates T1 signal hypointensity and T2/fluid attenuated inversion recovery (FLAIR) hyperintensity with occasional enhancement. They tend to develop in the frontal or temporal lobes; however, all lobes of the brain can harbor these tumours.2 Traditionally, LGG pathology was described in terms of histological appearance. With the updates to WHO 2021 classification for central nervous system tumours, LGG pathology is defined not only by the histology, but also by the molecular/genetic makeup of the tumour.3 The most important genetic aberration that defines adult-type diffuse LGG is mutations in the isocitrate dehydrogenase (IDH) gene.4 IDH has two isoforms, IDH1 in the cytoplasm and IDH2 in the mitochondria, with the most common mutation being the IDH1 mutation in the 132 codon from arginine to histidine (Figure 1).5,6 The normal IDH enzyme is part of the Krebs cycle and converts isocitrate to α-ketoglutarate. When the IDH1/2 mutant enzyme is present, α-ketoglutarate is converted to 2-hydroxyglutarate, the oncometabolite responsible for gliomagenesis.7 The observation is that all glial tumours with IDH mutations have an improved prognosis compared with those with IDH wildtype genotype, and this was the key motivation to make these specific categories in WHO 2021 classification system. Adult-type LGGs WHO grade 2 are currently described as oligodendroglioma, IDH-mutant, and 1p/19q-deleted and astrocytoma, IDH-mutant. 1p/19q co-deletion involves complete deletion of the short arm of chromosome 1 (1p) and the long arm of chromosome 19 (19q) and is specific to oligodendrogliomas (Table 1). Moreover, it is a predictive and prognostic biomarker in patients with oligodendroglioma as patients respond better to combinatorial chemotherapy and have a better prognosis.8,9
Figure 1: Mutant IDH lead to conversion of α-ketoglutarate to production of 2-HG, a oncometabolite, causing gliomagenesis
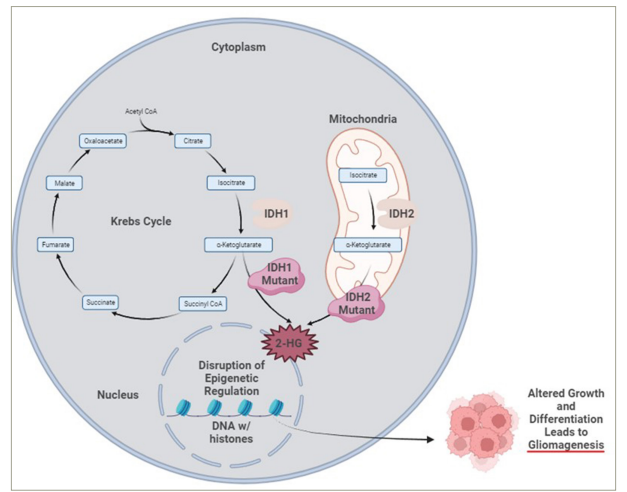
CoA = coenzyme A; IDH1 = isocitrate dehydrogenase 1; IDH2 = isocitrate dehydrogenase 2; 2-HG = 2-hydroxyglutarate. Created with BioRender.com.
Table 1: Criteria for low-grade glioma diagnosis in adults (per World Health Organization 2021 guidelines3)
|
| IDH1 mutation or IDH2 mutation | IDH1 mutation or IDH2 mutation |
| Associated mutations | ATRX retained, 1 p/19q co-deletion | ATRX mutant, TP53 mutation, 1 p/19q intact |
| Integrated WHO 2021 diagnosis | Oligodendriglioma, IDH-mutant, 1 p/19q co-deleted, WHO grade 2 | Astrocytoma, IDH-mutant, WHO grade 2 |
ATRX = alpha-thalassemia/mental retardation syndrome X-linked; IDH1 = isocitrate dehydrogenase 1; IDH2 = isocitrate dehydrogenase 2; IDH = isocitrate dehydrogenase; WHO = World Health Organization .
Table 2: Active and future clinical trials utilizing IDH-mutant inhibitors
| Trial name (ClinicalTrial.gov identifier) | Compound (route) | Mechanism | Study population | Phase | Estimated completion date |
| A Study of HMPL-306 in Advanced Solid Tumors With IDH Mutations (NCT04762602)27 | HMPL-306 (oral) | Dual IDH1/2-mutant inhibitor | Advanced solid tumours | Phase I | 30 September 2023 |
| A Study of DS-1001b in Patients With Chemotherapy- and Radiotherapy-Naive IDH1 Mutated WHO Grade II Glioma (NCT04458272)28,29 | DS-1001b (oral) | IDH1-mutant inhibitor | Grade 2 glioma | Phase II | 30 June 2026 |
| ViCToRy: Vorasidenib in Combination With Tumor Specific Peptide Vaccine for Recurrent IDH1 Mutant Lower Grade Gliomas (ViCToRy) (NCT05609994)30 | Vorasidenib (oral) and peptide vaccine (SC) | IDH1/2-mutant inhibitor and peptide vaccine to R132H IDH1 mutation | Lower grade glioma | Phase I | August 2027 |
| Study of Vorasidenib and Pembrolizumab Combination in Recurrent or Progressive Enhancing IDH-1 Mutant Astrocytomas (NCT05484622)31 | Vorasidenib (oral) and pembrolizumab (IV) | IDH1/2-mutant inhibitor and PD-1 inhibitor | Recurrent mutant astrocytoma | Phase I | 30 August 2027 |
| Ivosidenib (AG-120) With Nivolumab in IDH1 Mutant Tumors (NCT04056910)32 | Ivosidenib (oral) and nivolumab (IV) | IDH1-mutant inhibitor and PD-1 inhibitor | Gliomas and advanced solid tumours | Phase II | 31 May 2026 |
| Safusidenib Phase 2 Study in IDH1 Mutant Glioma (NCT05303519)33 | Safusidenib (oral) | IDH1-mutant inhibitor | Recurrent glioma | Phase II | 31 July 2027 |
IDH1 = isocitrate dehydrogenase 1; IDH2 = isocitrate dehydrogenase 2; IV = intravenous; PD = programmed cell death protein 1; SC = subcutaneous; WHO = World Health Organization .
Treatment of adult-type diffuse LGG before IDH mutation inhibition
Treatment of IDH-mutant adult-type diffuse LGG requires a multimodal approach with contributions from neurosurgery, neuropathology, radiation oncology, neuro-oncology, neurology and medical oncology. The first step in treatment is to determine the proper diagnosis. Classically, this determination was made by histology obtained by biopsy or craniotomy, but now providers are using molecular/genetic testing to determine the correct diagnosis. Providers can obtain this testing with in-house efforts from neuropathology or next-generation sequencing platforms. While variability exists in histopathology laboratories based on proprietary practices, in general, when a tumour is determined to be glial in origin, commercial antibody testing is performed for mutant IDH1 specific for the R132H mutation.10 If this test is negative, then it prompts more in-depth testing using next-generation sequencing. It is key to note that providers need to be aware that non-canonical mutations in IDH1 occur, along with IDH2 mutations. In special cases, DNA tumour methylation analysis is utilized, but it is not widely available.11
Greater extent of surgical resection is associated with improved prognosis in a systemic meta-analysis and is more attainable given the improved technology using functional mapping and awake craniotomy techniques.12,13 Moreover, uncontrolled studies point out that early initial resection is associated with improved prognosis over initial biopsy only.14
After the best possible safe surgery, recommendations for IDH-mutant adult-type diffuse LGG can include observation, radiation therapy with concurrent and adjuvant chemotherapy and consideration of clinical trials. This strategy is highlighted in the guidelines from the National Comprehensive Cancer Network (NCCN), European Association of Neuro-Oncology (EANO) and the Korean Society of Neuro-Oncology (KSNO).15 Consideration of observation after surgery with delay of radiation therapy is supported by the critical EORTC 22845 randomized clinical trial (Early versus delayed radiotherapy for low-grade astrocytoma and oligodendroglioma in adults) that randomized patients with LGG to either early radiation therapy or delayed radiation therapy.16 In this study of 314 patients with LGG randomized 1:1 to either early radiation therapy or delayed radiation therapy, results demonstrated that early radiation therapy improved progression-free survival, but there was a similar median overall survival for both groups (7.4 years for early radiation therapy versus 7.2 years for delayed radiation therapy, hazard ratio [HR] 0.97).
At the time of disease progression, patients are generally recommended to have radiation therapy with concurrent and adjuvant chemotherapy. In both the NCCN and KSNO guidelines, they stratify patients by high- and low-risk, in terms of recommendations for initial treatment after surgery.15 Factors associated with a higher risk of tumor progression include age >40 years and surgery less than gross total resection. The RTOG 9802 trial (Observation or radiation therapy with or without combination chemotherapy in treating patients with low-grade glioma; ClinicalTrials.gov identifier: NCT00003375) delineated the role of chemotherapy for patients with high-risk LGG, showing that the addition of combinatorial chemotherapy with procarbazine, lomustine and vincristine (PCV) to radiation therapy improved progression-free and overall survival compared with radiation therapy alone.17 In RTOG 9802, 251 patients with high-risk LGG were randomized to receive either radiation therapy alone or radiation therapy with PCV. Median progression-free survival was improved significantly in the combination group (RT + PCV) at 10.4 years when compared with the RT alone group at 4.0 years (HR=0.50, p<0.001). Median overall survival was superior in the combination group (RT + PCV) at 13.3 years compared with the RT alone group at 7.8 years (HR=0.59, p=0.003). Tolerability of PCV is challenging, as it can lead to cytopenias, fatigue and other toxicities, so temozolomide (TMZ), an oral alkylating agent US Food and Drug Administration (FDA)-approved for newly diagnosed glioblastoma IDH wildtype WHO grade 4, is commonly prescribed instead by providers.18
Many assumptions are made about how to approach the treatment of LGG, and these arise from clinical trial outcomes from newly diagnosed high-grade glioma studies. If avoiding toxicity is a goal in treating LGG, then delaying or eliminating the need for radiation therapy should be considered. Radiation therapy can cause significant cognitive dysfunction and lead to short- and long-term deficits.19 Douw and colleagues evaluated long-term LGG survivors who received radiation therapy and demonstrated a progressive decline in cognitive abilities.20 Additionally, these cognitive deficits were correlated with radiographic findings of white matter damage and cortical atrophy20. Better therapeutics for IDH-mutant adult-type diffuse LGG are needed to avoid the toxicities seen with conventional chemotherapy and radiation therapy. All in all, paradigms to treat IDH-mutant adult-type diffuse LGG deserve to be tailored to the specifics of the disease itself and should not extrapolated from treatment for high-grade gliomas such as glioblastoma IDH wildtype.
INDIGO trial: Vorasidenib in IDH-mutant LGG
An approach to treat LGG by targeting the tumour-specific mutation in the IDH1 and IDH2 genes has shown promise, based on recent results from a study by Mellinghoff et al.21 Vorasidenib is an oral, highly brain-penetrant inhibitor of mutant IDH1 and IDH2. In a phase I study of vorasidenib in recurrent and progressive glioma, vorasidenib was well-tolerated and safe, and median progression-free survival in patients with non-enhancing glioma was 36.8 months (95% confidence interval [CI], 11.2–40.8 months). Based on this study and a perioperative clinical trial that included vorasidenib, the INDIGO study (Study of vorasidenib [AG-881] in participants with residual or recurrent grade 2 glioma with an IDH1 or IDH2 mutation; ClinicalTrials.gov identifier: NCT04164901) was developed, and the dose of 40 mg orally once a day was determined to be the recommended phase III dose.22,23 In this pivotal phase III study, 331 patients with IDH-mutant LGG were enrolled and randomized in a blinded fashion to receive either placebo or vorasidenib. Key inclusion criteria included that prior treatment was limited to surgery only, and surgery should have occurred 1–5 years ago, and there had to be measurable non-enhancing disease of >1 cm and <1 cm enhancing disease. The primary endpoint was progression-free survival, and the key secondary endpoint was time-to-next intervention. The desired goal was to extend both of these endpoints particularly to delay the need for radiation therapy, further surgery, or initiation of chemotherapy. Patients receiving vorasidenib exhibited significantly longer progression-free survival than patients receiving placebo (27.7 months versus 11.1 months, HR 0.39, p<0.001). Time-to-next intervention was extended in the vorasidenib arm compared with the placebo arm in a highly significant manner (not reached at the time of analysis versus 17.7 months, HR 0.26, p<0.001). Given the success of these results, the study was concluded after an interim analysis, and patients receiving placebo were allowed to cross over to the vorasidenib arm. Vorasidenib was highly tolerated in this patient population, with only 3% of patients discontinued during the study. There was a higher incidence of elevated liver function tests in the vorasidenib arm, so care should be taken to monitor liver function tests appropriately and regularly. During the INDIGO clinical trial, liver function tests were monitored on day 1 and day 15 of the first two cycles and then on day 1 for all subsequent cycles (monthly) as long as the liver function tests remained within normal limits. With these results, vorasidenib is primed to become a critical therapeutic in treating newly diagnosed IDH-mutant LGG.
The INDIGO study enrolled patients from North America, Europe and Israel. A majority of patients were from North America (n=193). When comparing patients from North America, Europe, and Israel, there appears to be some suggestion of increased benefit in patients with IDH-mutant LGG in North America (HR=0.34) compared with patients in Europe (HR=0.54) and Israel (HR=0.45). Given the smaller number of patients enrolled in Europe and Israel, it is challenging to draw more significant conclusions. Still, these observations provide a rationale for exploring outcomes of vorasidenib in other populations on different continents, including South America, Asia, and Africa.
Before vorasidenib, ivosidenib (AG-120, Tibsovo®, Servier Pharmaceuticals, Paris, France), which is FDA-approved for both newly diagnosed and recurrent/refractory acute myelogenous leukemia, was the first-in-class oral small-molecule inhibitor of mutant IDH enzyme that blocks the conversion of α-ketoglutarate to 2-hydroxyglutarate.24,25 In a phase I dose-escalation study, ivosidenib had clinical activity in patients with recurrent IDH1-mutant non-enhancing glioma with a median progression-free survival of 13.6 months (95% CI, 9.2–22.2 months), and they were able to achieve stable disease in 35 patients (85.7%).26 When and how ivosidenib will be used in patients with brain tumours is limited by its poorer brain penetrance compared with vorasidenib, which can more easily cross the blood–brain barrier. One cannot discount the apparent usefulness of ivosidenib given that it appears to have similar efficacy in tumour response and reduction of 2-hydroxyglutarate as determined in the perioperative study comparing vorasidenib and ivosidenib in patients with recurrent IDH1-mutant LGG.23 Where ivosidenib fits into the future treatment regimens for IDH1-mutant glioma remains to be determined.
Next steps for inhibitors of IDH mutations
The results from the INDIGO study in newly diagnosed IDH-mutant LGG will undoubtedly change how patients with these tumours are treated. Vorasidenib is on track for FDA approval for this patient population. Moreover, this study highlights the recommendations from the WHO 2021 classification system for central nervous system tumours; obtaining molecular/genetic information for glial tumours is critical, and genotype status for IDH1 and IDH2 is paramount for prognostic and driving treatment decisions.
Three questions stand out from this randomized clinical trial. First, what is the optimal timing of these agents? For this study, patients were naïve to prior chemotherapy and radiation therapy, and only had prior surgery. Additionally, patients had to have measurable non-enhancing disease, but <1 cm of enhancing disease. Extrapolations from prior clinical trials, such as EORTC 22845 and RTOG 9802, that did not have the technology available for IDH genotype testing, are currently challenging to make for IDH-mutant adult-type diffuse glioma. One implication is that the extent and timing of surgical resection might not hold the same prognostic value in patients with IDH-mutant adult-type diffuse glioma. Time will tell how we can best use IDH-mutant inhibitors. One can posit that future clinical studies in newly diagnosed IDH-mutant LGG could include exploring the role of IDH-mutant inhibition at the time of diagnosis, perhaps even before definitive surgical resection.
Regarding the optimal timing of when to start IDH-mutant inhibitors, one can imagine that initiation of therapy could be different for patients depending on histological type (oligodendroglioma versus astrocytoma), location (non-eloquent versus eloquent), and extent of surgical resection (gross total resection versus subtotal resection versus biopsy). One can posit that healthcare providers of patients with more favorable factors, such as oligodendroglioma histology, location in non-eloquent regions and greater extent of surgery, might consider waiting before initiating therapy. On the other hand, providers with patients with less favorable factors, such as astrocytoma histology, location in eloquent regions, and suboptimal surgical resection, might favor the initiation of IDH-mutant inhibitors. Other factors to consider in initiating IDH-mutant inhibitors might include the need to preserve fertility and mitigate side effects, despite the favourable safety profile of vorasidenib.
Next, can we translate these results from INDIGO to our newly diagnosed patients with high-grade glioma with IDH mutations? Similar to how researchers and providers took the observations from WHO grade 3–4 glioma studies to guide therapeutic regimens in IDH-mutant LGG, vorasidenib will likely be utilized in the research and treatment of IDH-mutant high-grade gliomas. Upcoming and active studies are looking at low-grade and high-grade glioma populations (Table 2). Future trials will need to explore the role of IDH-mutant inhibitors during radiation therapy or during adjuvant therapy for higher-grade IDH-mutant tumours. Future endeavours will only add to the evidence and provide more options for all patients with IDH-mutant glioma.
Finally, how does vorasidenib impact the quality-of-life outcomes in patients with IDH-mutant glioma? INDIGO explored the quality of life, cognition and seizure outcomes in these patients, and the results will be published in the future. Patients with IDH-mutant adult-type diffuse LGG experience significant symptoms and impairments in their quality of life.34 Common issues these patients face include seizures, fatigue, cognitive dysfunction, headaches, sleep disturbances and mood disorders.35 Interprofessional, multidisciplinary teams from neuro-oncology, neurology, neurosurgery, neuropsychology, psychiatry, radiation oncology, physical therapy, occupational therapy and other subspecialties are vital to supporting these patients through their disease trajectory. In particular, seizures are more common in patients with IDH-mutant glioma. Work from Chen and colleagues showed that preoperative seizures were more common in patients with IDH-mutant glioma (59–74%) compared with patients with IDH-wildtype glioma (18–34%).36 While this association could be secondary to the location in epileptically prone areas (frontal and temporal lobes), they found that exogenous 2-hydroxyglutarate promoted excitatory activities in cultured neurons. The presence of 2-hydroxyglutarate in IDH-mutant tumours stimulates local neurons towards stimulatory activity that leads to seizures. Researchers will have an opportunity with the data from INDIGO to explore these particular issues in this patient population. Meaningful design of the relevant patient-reported outcomes and integration into future clinical trials are warranted.
This randomized clinical trial represents a practice-changing step in treating IDH-mutant diffuse adult-type LGG by utilizing a targeted approach to IDH mutations. Active and upcoming clinical studies utilizing IDH-mutant inhibitors are ongoing and are likely to establish when and how providers will use IDH-mutant inhibitors in treating all grades of glioma that have an IDH mutation (Table 2).27–33