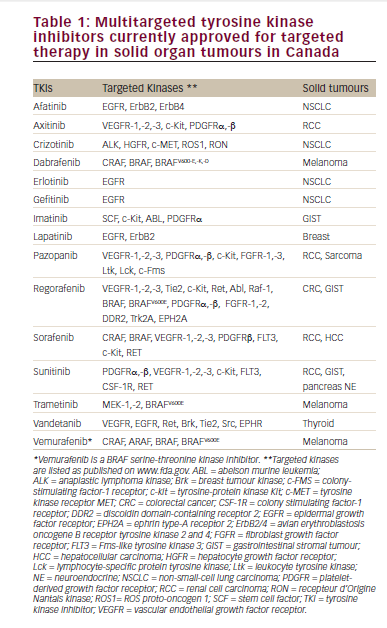
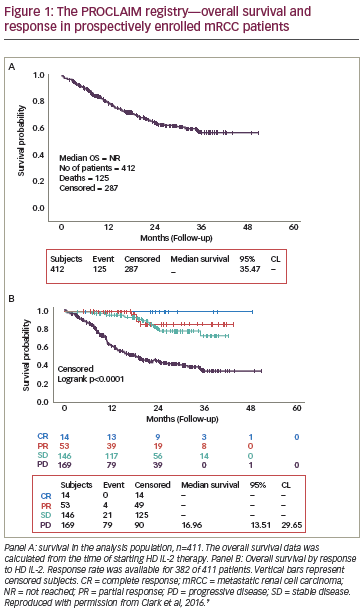
Renal cell carcinoma (RCC) accounts for 3% of all cancers.1,2 Approximately 30% of patients present with advanced and/or metastatic RCC (mRCC) at the time of diagnosis or develop metastases over time.3 Additionally, one-third of patients treated surgically for localised disease subsequently relapse and develop metastatic disease.3 The prognosis for patients with metastatic disease is poor, with a five-year survival rate of ~10%.4
mRCC is resistant to conventional chemotherapy, radiotherapy and hormonal therapies, and until recently the mainstay of treatment for patients with mRCC was cytokine therapy with interleukin-2 (IL-2) or interferon-α (IFN-α).5 However, these treatments have limited efficacy in the majority of mRCC cases and are associated with substantial toxicities.6 Recent developments in our understanding of the molecular biology of RCC have identified several pathways associated with the development of the disease, and the introduction into clinical practice of targeted agents that inhibit these pathways has led to dramatic improvements in prognoses for mRCC patients. However, several questions remain unanswered in terms of the optimal use of these agents, including the most effective sequences of therapy and the relative efficacy of combination versus sequential single-agent therapy.
This article discusses the current approaches to the management of mRCC and reviews strategies for potential treatment optimisation, including the use of targeted agents in sequence and combination and the integration of targeted treatments in the neoadjuvant and adjuvant settings.
Immunotherapy
Until recently, immunotherapy with IFN-α and IL-2 was the only effective treatment available for mRCC and represented the standard of care in this setting, although cumulative experience with these therapeutics suggests only modest benefits.6 The objective response rate (ORR) with IFN-α therapy is approximately 15%,7 and a significant but modest survival advantage for IFN-α compared with nonimmunotherapy was reported in two randomised trials and a meta-analysis.6,8,9 An ORR of 17–27% was reported with high-dose IL- 2 therapy in mRCC patients,10 with durable complete responses observed in approximately 5% of patients.11 However, the benefit of both of these agents is limited to select patients with good prognosis for therapy;12,13 a recent trial showed no benefit from IFN-α therapy in mRCC patients with intermediate risk.14 Furthermore, the substantial toxicities observed with these cytokines limits their use in this setting.15,16 Therefore, the current role for cytokine therapy in mRCC is evolving and unclear. Current investigative efforts are being targeted towards combination therapy and patient selection. For example, carbonic anhydrase IX expression has been associated with a higher ORR with IL-2 in retrospective analyses, identifying it as a potential independent prognostic marker in patients with mRCC, although its utility in this setting has been questioned.17-19
Nephrectomy
Cytoreductive nephrectomy followed by immunotherapy is recommended for patients with mRCC who have potentially resectable primary tumours. This is based on the results of two clinical studies that investigated the benefit of cytoreductive nephrectomy followed by cytokine therapy. These prospective, randomised trials from the Southwest Oncology Group (SWOG) and the European Organisation for the Research and Treatment of Cancer (EORTC) evaluated nephrectomy followed by IFN-α compared with IFN-α alone.20,21 In both studies, overall survival was significantly longer when nephrectomy was part of the treatment strategy (SWOG 11.1 versus 8.1 months, EORTC 17 versus seven months). A combined analysis of these studies revealed a median survival advantage of 5.8 months when nephrectomy was followed by IFN-α (p=0.002).22 Patients most likely to benefit from cytoreductive therapy are those with good performance status, good prognostic features and limited metastatic burden.23 However, the treatment paradigm for mRCC is evolving and these studies pre-date the advent of targeted therapies. The potential survival benefits of nephrectomy in the context of targeted agents remain to be determined. Ongoing studies, such as the European phase III Clinical Trial to Assess the Importance of Nephrectomy (CARMENA; clinicaltrials.gov identifier NCT00930033), are exploring the role of nephrectomy in patients exposed to targeted treatment.
Targeted Therapy
An increased understanding of the molecular biology of mRCC has resulted in the identification of several viable therapeutic targets and novel agents that inhibit these pathways, leading to major improvements in the treatment options for mRCC (see Table 1). These agents constitute two mechanistic classes: angiogenesis inhibitors targeting the vascular endothelial growth factor (VEGF) ligand (e.g. bevacizumab) or VEGF receptors (VEGFR) (such as the multikinase inhibitors sunitinib, pazopanib and sorafenib), and inhibitors of the mammalian target of rapamycin (mTOR) signalling pathway (everolimus, temsirolimus). The hypervascularity observed in RCC tumours is frequently driven by the inactivation of the von Hippel- Lindau (VHL) gene, particularly in tumours with clear-cell histology, the most prevalent histological class of RCC. This leads to overexpression of hypoxia-inducible factor-1α (HIF), a transcription factor that regulates the expression of pro-angiogenic factors such as VEGF, providing a rationale for targeting this pathway.24,25 The mTOR pathway is a central component of multiple tumour-promoting intracellular signalling pathways,26 including HIF expression, and activation of this pathway has been documented in mRCC.27
Multikinase Inhibitors
Sunitinib
Sunitinib is a tyrosine kinase inhibitor (TKI) that targets VEGFR2, platelet-derived growth factor receptor (PDGFR), FMS-like tyrosine kinase 3 (FLT-3) and c-KIT.28,29 Sunitinib is administered as an oral dose of 50mg once a day for four weeks followed by a two-week break. Two multicentre phase II studies of sunitinib in mRCC patients who had failed previous cytokine therapy demonstrated similar results: partial responses were reported in 40 and 34% in the two studies, respectively, and median progression-free survival (PFS) was 8.8 and 8.3 months, respectively.30,31 Fatigue was the most common adverse event in both studies.
The favourable results of these phase II studies led to a phase III trial of sunitinib versus IFN-α as first-line therapy.32 This study recruited 750 treatment-naïve patients to repeated six-week cycles of sunitinib or IFN-α. Over 90% of patients recruited to the study had a favourable or intermediate prognosis and 90% had undergone prior nephrectomy.
A significant improvement in median PFS (the primary end-point) was observed in the sunitinib arm compared with IFN-α (11 versus five months, hazard ratio [HR] 0.42; p<0.001), and sunitinib-treated patients also experienced significant improvements in ORR (31 versus 6%; p<0.001). Adverse events were similar in the two groups, except that more patients (5–10%) in the sunitinib group experienced grade 3 or 4 diarrhoea or hypertension, whereas grade 3 or 4 fatigue was more common in IFN-α-treated patients. An updated analysis from this study demonstrated that the median overall survival was longer in the sunitinib group than the IFN-α group (26.4 versus 21.8 months, respectively, HR 0.82; p=0.051), although the statistical significance was marginal.33 However, the survival end-point may have been confounded by cross-over treatments and is potentially underestimated.
Based on these data, sunitinib is approved multinationally for the first-line treatment of mRCC and is considered a reference standard of care in the first-line setting for patients at favourable or intermediate prognostic risk. Based on the antitumour activity observed with sunitinib in the second-line setting,30,31 the US National Comprehensive Cancer Network (NCCN) Kidney Cancer Guidelines also recommend sunitinib treatment following cytokine therapy (category 1 recommendation) or after prior TKI therapy (category 2A recommendation).34 Studies investigating the scheduling of sunitinib and the combination of this agent with other active compounds are ongoing, although results from a phase I evaluation of sunitinib plus IFN-α in first-line mRCC patients were disappointing.35
Sorafenib
Sorafenib is an oral TKI that targets the receptor tyrosine kinases VEGFR2, VEGFR3, FLT3, c-KIT and PDGFR and the non-receptor serine/threonine kinase RAF1, a major protein of the mitogen-activated protein kinase (MAPK) pathway.36 Therefore, sorafenib is able to target both tumour angiogenesis and proliferation. Based on the results of a pivotal phase III randomised trial, sorafenib is recommended for the second-line treatment of mRCC following cytokine therapy.37 In this study, 903 patients who had either received or were ineligible for cytokine therapy were randomised to receive sorafenib (at a dose of 400mg twice daily) or placebo; this dose of sorafenib was previously shown to be effective in a randomised phase II trial in mRCC.38 A planned interim analysis of PFS in this phase III study showed a statistically significant advantage in the sorafenib arm (5.5 versus 2.8 months, HR 0.44; p<0.01). The ORR was also significantly higher in the sorafenib arm compared with placebo (10 versus 2%; p<0.001).37 As a consequence of these results, placebo patients were permitted to cross-over to sorafenib. A significant survival advantage was seen with sorafenib in a per-protocol analysis adjusting for cross-over (17.8 versus 14.3 months, HR 0.78; p=0.0287).39 Adverse events were generally manageable: diarrhoea, rash, fatigue and hand–foot skin reactions were the most common adverse events associated with sorafenib. Hypertension and cardiac ischaemia were rare serious adverse events that were more common in patients receiving sorafenib than in those receiving placebo. In the first-line mRCC setting, a randomised phase II study of 189 patients demonstrated no difference in median PFS with sorafenib treatment compared with IFN-α (5.7 and 5.6 months, respectively), although sorafenib-treated patients demonstrated greater rates of tumour size reduction, better quality of life and improved tolerability.40
Finally, two phase II studies investigating the combination of sorafenib and IFN-α in mRCC demonstrated an ORR of 19 and 33%, respectively, and an encouraging median PFS (seven and 10 months, respectively).41,42 However, increased toxicities with the combination regimen compared with either drug alone was a concern in both of these studies.
Pazopanib
Pazopanib is an inhibitor of several kinases, including VEGFR1, -2 and -3, PDGFR-α/β and c-KIT. This agent demonstrated monotherapy activity in RCC during phase I/II evaluation,43,44 paving the way for a phase III randomised, placebo controlled trial in treatment-naïve and cytokine-refractory mRCC patients (n=435).45 PFS (the primary endpoint) was significantly prolonged with pazopanib (oral dose of 800mg once daily) compared with placebo in the overall study population (median 9.2 versus 4.2 months, HR 0.46; p<0.001) and ORR was also significantly improved (30 versus 3%; p<0.001). PFS with pazopanib in the treatment-naïve subpopulation (11.1 versus 2.8 months in the placebo arm) was similar to that observed with sunitinib. Pazopanib demonstrated acceptable safety and tolerability and most adverse events related to pazopanib treatment were grade 1 or 2 and were clinically manageable. A possible criticism of this pivotal trial is that it was conducted with a placebo group instead of an active comparator from among the therapeutic options already approved for this indication. However, a phase III trial comparing pazopanib monotherapy with sunitinib in treatment-naïve mRCC patients is ongoing (clinicaltrials.gov identifier NCT00720941).
Anti vascular Endothelial Growth Factor Agents
Bevacizumab
Bevacizumab is a humanised monoclonal antibody that binds and neutralises circulating VEGF, thus preventing it from binding to its receptor. A controlled, double-blind phase III study (AVOREN) randomised 649 first-line mRCC patients to receive IFN-α (9MIU three times weekly) and bevacizumab (10mg/kg every two weeks) or IFN-α and placebo.46 Significant advantages in median PFS and ORR were reported for the bevacizumab arm compared with IFN-α alone (PFS 10.2 versus 5.4 months, HR 0.63; p=0.0001; ORR 31 versus 13%; p<0.0001), but the final analysis of the primary end-point of median overall survival demonstrated no significant improvement for the bevacizumab combination versus IFN-α (23.3 versus 21.3 months; HR 0.86, p=0.1291).47 These results were confirmed in the Cancer and Leukaemia Group B 90206 (CALGB 90206) open-label phase III study, which randomised 732 first-line mRCC patients to either bevacizumab plus IFN-α or IFN-α monotherapy without placebo control (median PFS 8.5 versus 5.2 months, HR 0.71; p<0.0001; ORR 25.5 versus 13.1%; p<0.0001).48 As seen with the AVOREN study, median overall survival favoured the bevacizumab plus IFN-α arm but did not meet the pre-defined criteria for significance (18.3 versus 17.4 months, HR 0.86; p=0.069).49 Grade 3–4 hypertension, anorexia, fatigue and proteinuria were more commonly observed in the bevacizumab plus IFN-α arm. Based on these data, bevacizumab plus IFN-α is recommended as a first-line therapy for patients with mRCC. However, both studies demonstrated that poor-prognosis patients were unlikely to benefit from this combination.
Mammalian Target of Rapamycin Inhibitors
Temsirolimus
Temsirolimus was the first inhibitor of mTOR kinase available for treating mRCC. Promising phase II results50,51 led to the initiation of a phase III trial in which 626 mRCC patients with previously untreated poor prognosis (according to the Memorial Sloan-Kettering Cancer Center [MSKCC] prognostic score)52 were randomised to receive temsirolimus (25mg intravenously [IV] once weekly), IFN-α (3MIU three times weekly) or a combination of both.53 Temsirolimus alone prolonged overall survival (median 10.9 versus 7.3 months, HR 0.73; p=0.008) and PFS (median 3.8 versus 1.9 months, HR 0.42; p<0.001) compared with IFN-α. Overall survival in the combination-therapy group did not differ significantly from that in the IFN group (HR 0.96; p=0.70). ORR did not differ significantly between the treatment groups, although the proportion of patients with stable disease for ≥6 months or an objective response (clinical benefit rate) was greater in temsirolimus-treated patients compared with IFN-α-treated patients (32.1 versus 15.1%; p<0.001). Rash, peripheral oedema, hyperglycaemia and hyperlipidaemia were more common in the temsirolimus group, whereas asthenia was more common in the IFN-α group. Fewer patients experienced serious adverse events in the temsirolimus group compared with the IFN-α group. This study demonstrated that temsirolimus is a suitable option for the treatment of poor-prognosis mRCC patients and is recommended as a first-line therapy in this setting.
Everolimus
Everolimus is another mTOR inhibitor that is available for the treatment of patients with mRCC. Unlike the intravenously administered temsirolimus, everolimus is orally active. Everolimus demonstrated encouraging antitumour activity in a phase II study of patients with previously treated mRCC.54 A subsequent phase III trial randomised 410 patients with mRCC whose disease had progressed on sunitinib, sorafenib or both (prior therapy with cytokines or bevacizumab was also allowed) to receive either everolimus (10mg once daily) or placebo.55 The trial was stopped early after a second interim analysis had shown a significant difference in the primary end-point of PFS between the two groups (median four versus 1.9 months, HR 0.30; p < 0. 0001). An updated analysis showed that this significant PFS benefit was maintained during an additional 4.5 months of blinded follow-up (median 4.9 versus 1.9 months, HR 0.33; p<0.001).56 This PFS benefit was maintained across all MSKCC riskclassification groups. The ORR with everolimus treatment was modest (1.8% in the updated analysis), suggesting that the effect of everolimus on PFS is the result of disease stabilisation. There was no significant difference in median overall survival between the arms (14.8 versus 14.4 months for everolimus and placebo, respectively, HR 0.87; p=0.162), although the survival results were likely to have been confounded by the majority of patients (80%) in the placebo arm who switched therapy following disease progression.56 According to a post hoc exploratory analysis correcting for this cross-over effect, the corrected placebo overall survival was 10 months, a difference of 4.8 months compared with the observed everolimus overall survival.56 Adverse events reported with everolimus treatment were mostly grade 1 or 2. The most common events were stomatitis, rash, fatigue or asthenia and diarrhoea. Based on these data, the latest European Association of Urology (EAU) and NCCN (category 1 recommendation) guidelines recommend everolimus for the second-line treatment of mRCC after prior treatment with VEGF TKI therapies.
Sequencing Targeting Agents
Sequential therapy with targeted agents could potentially further improve outcomes in patients with mRCC. A sequential strategy could allow a treatment regimen to be established whereby patients are maintained on a continuum of treatments without tumour progression. Furthermore, targeting different pathways through sequential therapy could offer benefits in terms of overcoming resistance to individual agents.
The first randomised phase III study that investigated sequential targeted therapy in mRCC compared the mTOR inhibitor everolimus versus placebo in patients who had progressed on sunitinib, sorafenib or both.55,56 The significant prolongation of PFS observed with everolimus compared with placebo in the overall study population (HR 0.33) was maintained in subgroups of patients who had previously received sunitinib (HR 0.34) or sorafenib (HR 0.25). A retrospective analysis of temsirolimus in patients previously treated with VEGF-targeted therapy suggested that this sequence was feasible and active, albeit with an increased frequency of adverse events compared with that seen with temsirolimus in previously untreated poor-risk patients.57 These data suggest that the development of cross-resistance between inhibitors of VEGF or mTOR pathways may be minimal. Furthermore, the distinct adverse event profiles of mTOR inhibitors and VEGFR TKI therapies suggest an absence of overlapping toxicities, which may prove advantageous when considering sequential strategies involving these two mechanistic classes of targeted agents.58
Several retrospective analyses support the benefit of sequential sorafenib and sunitinib in this setting, suggesting that the sequencing of both of these TKIs is not hampered by cross-resistance, despite partly blocking the same signalling pathways.59–62 However, results from prospective studies suggest that sorafenib has limited efficacy in patients who experience treatment failure with first-line sunitinib. A prospective study of 42 patients with mRCC refractory to either firstline bevacizumab or sunitinib was able to demonstrate a median PFS of 3.7 months with sorafenib as a second-line treatment, but no objective responses were observed.63 Furthermore, significant toxicities were seen in this study (58% of patients experienced a grade 3 adverse event). Data from another prospective study of 52 sunitinibrefractory mRCC patients suggested that sorafenib has modest efficacy (partial response rate of 9.6%) in patients who experience treatment failure with first-line sunitinib.64 Taken together, these prospective studies highlight potential problems of cross-resistance and overlapping toxicities between these agents, emphasising the need for further prospective clinical trials investigating the sequencing of sunitinib and sorafenib. An exploratory phase II study investigating the antitumour activity of sunitinib in 61 patients with bevacizumabrefractory mRCC demonstrated a PFS of 30.4 weeks and an ORR of 23%, suggesting a lack of cross-resistance between sunitinib and bevacizumab.65
Further ongoing studies should provide important insights into the sequencing of targeted therapies in mRCC. A phase III study is comparing the efficacy and safety of temsirolimus versus sorafenib as a second-line therapy in mRCC patients who have failed first-line sunitinib (Temsirolimus Versus Sorafenib as Second-Line Therapy in Patients With Advanced RCC Who Have Failed First-Line Sunitinib; clinicaltrials.gov identifier NCT00474786), and a phase II trial (Efficacy and Safety Comparison of RAD001 Versus Sunitinib in the First-line and Second-line Treatment of Patients With Metastatic Renal Cell Carcinoma [RECORD-3]; clinicaltrials.gov identifier NCT00903175) is comparing the efficacy and safety of the sequence of first-line everolimus followed by second-line sunitinib versus the reverse sequence in patients with mRCC. Preliminary evidence suggests a potential clinical benefit with VEGFR TKI treatment in selected mRCC patients refractory to VEGF-targeted therapies and mTOR inhibitors (i.e. a sequence of VEGFR TKI followed by mTOR inhibitor followed by VEGFR TKI), suggesting a potential for ‘re-sensitisation’ to VEGFR TKI following mTOR therapy and additional studies may be warranted in this patient population.66,67 Finally, following promising phase II results,68 the efficacy and safety of the investigatory VEGFR inhibitor axitinib compared with sorafenib is being evaluated in a phase III study (Axitinib As Second Line Therapy For Metastatic Renal Cell Cancer [AXIS]) of mRCC patients who have progressed on first-line treatment (including sunitinib, bevacizumab plus IFN-α, temsirolimus or cytokines).
Combination Therapy with Targeted Drugs
Combination regimens are being actively studied in mRCC with the hope of additionally increasing the efficacy of targeted therapies. Inhibition of multiple molecular pathways critical to tumour growth and progression has the potential for improving tumour regression compared with single agents and may prevent the development of clinical resistance;69 however, this approach may lead to an increase in toxicity.
Complete inhibition of the VEGF pathway by combining anti-VEGF therapy and VEGFR TKIs has met with limited success in early-phase clinical studies owing to the toxicity of these regimens. In a phase I study of bevacizumab plus sorafenib, partial responses were observed in 21 out of 46 mRCC patients (46%) and the median time to progression was 11.2 months. However, this combination required dose reduction of both agents and bevacizumab appeared to exacerbate certain sorafenib-related toxicities, including hand–foot syndrome.70 The combination of sunitinib and bevacizumab was shown to cause a high degree of hypertension and vascular and haematological toxicities in patients with mRCC, also raising doubts about this regimen in this setting.71
Combination regimens of VEGF-targeted agents and mTOR kinase inhibitors are being investigated in an effort to attain synergistic antitumour activity or to prevent the development of cross-resistance. The combination of standard doses of everolimus and bevacizumab has demonstrated efficacy and tolerability in both untreated mRCC patients and those previously treated with sorafenib in a phase II study; ORR was similar in both groups of patients (untreated 30%, previously treated 23%).72 Temsirolimus and bevacizumab in combination was active and tolerable at recommended doses in mRCC,73 although temsirolimus in combination with sunitinib or sorafenib was associated with excessive toxicity at the doses tested in phase I studies.74,75 Additional larger studies are needed to fully elucidate the efficacy and safety of combination regimens of targeted agents in mRCC.
Integrating Surgical and Targeted Therapies
Neoadjuvant Targeted Therapy
Neoadjuvant strategies that integrate surgical intervention with systemic therapy may hold promise as a treatment paradigm in mRCC.76 The utility of pre-operative systemic therapy for tumour downsizing has not been explored extensively owing to the rare objective responses to cytokine therapy. However, the efficacy of targeted agents in the metastatic setting has stimulated interest in their potential for the downsizing of primary tumours and improving the feasibility of subsequent nephrectomy. Indeed, preliminary studies have suggested the efficacy of neoadjuvant sunitinib in advanced RCC,77,78 including a complete histological remission following neoadjuvant treatment with this agent in a clinical case study.79
Prospective studies investigating neoadjuvant sunitinib in advanced RCC have shown only moderate tumour downsizing and in some cases only tumour stabilisation. In a study of 17 patients with advanced RCC treated with sunitinib, only four patients had a partial response and the mean tumour volume reduction was 31%, with one patient showing tumour progression.80 In another study, eight out of 19 patients (42%) with advanced RCC who were treated with neoadjuvant sunitinib showed primary tumour shrinkage with a median tumour size decrease of 24%.81 Ten patients (53%) experienced disease progression. Taken as a whole, the role of neoadjuvant targeted therapy in patients with locally advanced /mRCC is not well defined and requires further study.
Adjuvant Targeted Therapy
The risk of recurrence after nephrectomy is correlated to parameters such as tumour grade, ECOG performance status or tumour histological necrosis. High-risk renal tumours have a 35–65% risk of recurrence.82 Effective adjuvant therapy may therefore reduce the risk of relapse in these patients. However, no benefit has been obtained in studies of adjuvant immunotherapy in RCC,83,84 and adjuvant systemic treatment is not currently indicated for this setting. No data are currently available for the use of targeted agents in the adjuvant setting but several trials (Adjuvant Sorafenib or Sunitinib for Unfavorable Renal Carcinoma [ASSURE], Sunitinib Treatment Of Renal Adjuvant Cancer [S-TRAC] and A Phase III Randomised Double-Blind Study Comparing Sorafenib With Placebo in Patients With Resected Primary Renal Cell Carcinoma at High or Intermediate Risk of Relapse [SORCE]; clinicaltrials.gov identifiers NCT00326898, NCT00375674 and NCT00492258, respectively) are ongoing to evaluate the use of sunitinib or sorafenib as adjuvant therapies in intermediate- and/or high-risk surgically resectable RCC.
Conclusions
Clinical studies have validated the utility of targeted inhibitors of the VEGF and mTOR pathways for treatment of mRCC, significantly improving the prognosis for many patients. Furthermore, the generally favourable toxicity profile of these agents renders them more appealing than previously used cytokine therapies. Ongoing research is focusing on identifying the optimal sequence and combinations of these agents, together with the potential role for targeted agents in the adjuvant and neoadjuvant settings.
Based on the efficacy and safety data from phase III trials and the approval status of targeted therapies for mRCC, sunitinib or bevacizumab plus IFN-α are considered first-line treatments in patients with good or intermediate prognosis, with temsirolimus the recommended first-line treatment in patients with poor
prognosis. Sorafenib is recommended for patients whose disease has progressed after cytokine therapy (second-line sunitinib therapy is also an option), whereas everolimus is recommended for patients whose disease has progressed after TKI therapy. It should be emphasised that these recommendations apply to patients with clear-cell RCC (the most prevalent histology). Except for temsirolimus, most clinical trials of targeted therapies enrolled patients with clear-cell RCC exclusively. This is not surprising as the biology of VEGF expression as a result of VHL mutation applies exclusively to clear-cell RCC and therefore the activity of VEGF-targeting agents is expected to be greater in patients with this histology. Enrolment in clinical trials is the preferred strategy for the minority of RCC patients with non-clear-cell histology (NCCN guidelines). ■