Cervical cancer (CC) is the fourth most common female malignancy, with 640,127 new cases and 341,831 deaths in 2020 worldwide. Despite the introduction of screening programmes and vaccination campaigns, CC still represents one of the leading causes of cancer-related death in women.1
The appropriate treatment strategy is determined according to the stage of disease at diagnosis. Patients with early-stage and locally advanced disease are treated with potentially curative intent with surgery and exclusive concomitant chemo-radiotherapy, respectively. In contrast, patients with metastatic, recurrent and persistent disease have a dismal prognosis, with a 5-year survival rate of less than 20%.2 In these patients, platinum-based chemotherapy represents the standard of care. The Gynecologic Oncology Group protocol 240 (GOG-240) trial reported a significant improvement in overall survival (OS) in patients treated with chemotherapy plus bevacizumab compared with chemotherapy alone (16.8 months versus 13.3 months, hazard ratio 0.77, 95% confidence interval [CI] 0.62–0.95; p=0.0068).3,4 Unfortunately, effective second-line treatments are lacking for patients who progress on first-line chemotherapy. Several cytotoxic agents, including vinorelbine, topotecan, gemcitabine and pemetrexed, have been tested, with low reported response rates ranging between 4.5% and 20%, as well as poor median OS of about 5 months.5 In this scenario, the evaluation of new, more active treatment strategies represents an open field of research and an urgent unmet clinical need.
In recent years, clinical studies have focused on the possible role of immunotherapy in recurrent or metastatic CC, and in 2018, the US Food and Drug Administration (FDA) approved pembrolizumab based on the KEYNOTE-158 study6 for advanced/recurrent CC expressing programmed cell death-1.
A new perspective: Antibody–drug conjugates
Antibody–drug conjugates (ADCs) are a novel class of agents consisting of three main components: antibody, linker and payload.7 The monoclonal antibody interacts with an antigen expressed on tumour cells, and with the help of the linker, cytotoxic agents are conveyed inside the cell and degraded by lysosomes. Once free, the cytotoxic effector can perform its function, causing cell death through several mechanisms. The monoclonal antibody recognizes an antigen predominantly expressed on tumour cells relative to normal cells in order to maximize efficacy while reducing toxicity. The monoclonal antibodies are mainly represented by the immunoglobin (IgG) subtype engineered to join payload molecules. A stable attachment is granted by cysteine variations or mutations that are able to modify electric charge and create stable bonds between immunoglobulins and cytotoxic agents. The drug:antibody ratio is defined as the number of payloads attached to IgG; however, a higher drug:antibody ratio does not necessarily confer enhanced efficacy because it could interfere with the pharmacokinetics of the ADC.8 The antigens targeted in gynaecological cancers include folate receptor α, sodium-dependent phosphate transport protein 2B, tissue factor (TF), mesothelin and mucin 16.
The payload is usually represented by small cytotoxic molecules measuring 300–1,000 daltons; agents with significant efficacy are auristatin and maytansine, both potent microtubule inhibitors.9 Among auristatins, monomethyl auristatin E (MMAE/vedotin) has shown a bystander effect on neighbouring cells owing to its ability to cross cell membranes. Monomethyl auristatin F (MMAF/mafodotin), however, did not show the same activity. Among maytansines, we distinguish emtansine, mertansine, soravtansine and ravtansine.
The linker is fundamental to the stability of ADCs in the bloodstream, avoiding any inappropriate release with subsequent major adverse effects. The linkage between the main components can be cleavable or non-cleavable. Cleavable linkers are split by enzymatic degradation and hydrolysis reactions, whereas non-cleavable linkers are divided within lysosomes, together with the antibody backbone.
The first-in-class ADCs were approved by the FDA for the treatment of haematological malignancies: brentuximab vedotin for Hodgkin lymphoma and anaplastic large cell lymphoma, gemtuzumab ozogamicin for acute myeloid leukaemia, and inotuzumab ozogamicin for B-cell acute lymphoblastic leukaemia.10 Concerning solid tumours, only ado-trastuzumab emtansine is approved for the treatment of advanced human epidermal growth factor receptor 2-positive breast cancer.11
Among gynaecological malignancies, several clinical trials have investigated the role of ADCs. The activity of mirvetuximab soravtansine has been explored in ovarian cancer and endometrial cancer, while anetumab ravtansine (DMOT4039A) and sofituzumab vedotin (DMUC5754A) have both been studied in ovarian cancer. The efficacy and safety of tisotumab vedotin (TV) have been investigated in ovarian, endometrial and cervical cancers. The aim of the current review is to describe the role of TV in metastatic and recurrent CC, and to provide an overview of published and ongoing clinical trials in this setting.
Methods
Following the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, a search of PubMed and Scopus databases was performed to identify publications up to 25 April 2021. The following combination of Medical Subject Headings (MeSH) terms was used in both databases: “cervix cancer”, “cervical cancer”, “solid tumor”, “tisotumab vedotin”, “antibody-drug conjugated”. We included published clinical trials without limitations on the publication date. We also searched ClinicalTrials.gov for ongoing trials. We excluded reviews, abstracts and articles not written in English.
Tisotumab vedotin: An overview
TV is an ADC targeting TF, a transmembrane glycoprotein mainly known as the primary initiator of the extrinsic coagulation pathway.12
TF is physiologically expressed on subendothelial cells (fibroblasts, pericytes and smooth muscle cells), and the exposition, secondary to injury, promotes the activation of blood-circulating factor VII, leading to coagulation.13 Moreover, the TF–factor VIIa complex mediates an intracellular signalling cascade by activating protease-activated receptor 2, leading to the production of pro-angiogenetic factors and cytokines.12 Alongside its function as a coagulation promoter, TF also plays a role in oncogenesis, promoting tumour cell survival, tumour growth and metastasis. TF is overexpressed in several solid tumours,14 including in gynaecological cancers such as CC (100%),15 endometrial cancer (14–100%)16 and ovarian cancer (75–100%),17 where it is associated with poor prognosis.18
Owing to its aberrant expression on tumour cells, TF is a target for ADC therapy. After binding with TF, TV releases its microtubule-disrupting agent MMAE inside the cell, causing cell cycle arrest and apoptotic cell death.19,20 Moreover, as reported in preclinical studies, MMAE can diffuse across the cell membranes of neighbouring tumours, where it performs the same cytotoxic process.
In addition to its direct cell death activity, the cytotoxic effect of TV is also fulfilled through an indirect mechanism. The drug can activate the immune system through the FcgRIII expressed on natural killer cells, activating antibody-dependent cellular cytotoxicity (ADCC) and inducing immunogenic cell death; this process also results in the release of neoantigens, which play an important role in the specific adaptative immune response.21
TV was selected from a panel of six human TF-specific antibodies (HuMab-TF) because of its reduced interference with the pro-coagulant activity of TF.20
Tisotumab vedotin in cervical cancer
Preclinical evidence
The interaction between TF and ADC was firstly evaluated in preclinical studies. Breij, et al. studied the activity of monoclonal antibodies conjugated with cytotoxic agents directed against TF both in vitro and in vivo.20
Human tumour cells from pancreatic (AsPC-1, BxPC-3 and HPAF-II), colorectal (HCT-116), breast (MDA-MB-231), ovarian (SK-OV-3 and TOV-21G) carcinoma, and epidermal adenocarcinoma (A431) were collected and tested for TF expression. QIFIKIT® (Dako, Glostrup, Denmark) analysis was performed using a mouse-derived antibody. In addition, a codon-optimized construct was generated for TF expression and transferred into HEK-293F cells or NS0 cells. Human IgG1k TF antibodies (HuMab) were created through the immunization of HuMab mice. Three types of monoclonal antibodies (TF-011, TF-098 and TF-111) were conjugated with cytotoxic agents MMAE and MMAF. A protease-cleavable valine citrulline dipeptide was used for MMAE linking, whereas a non-cleavable maleimidocaproyl-containing linker was necessary for MMAF. The final result was an ADC.20
The binding between the ADC and TF was evaluated with flow cytometry. A negative interaction between HuMab-TF and physiological coagulation was also excluded: citrated human blood from healthy volunteers was incubated with humanized immunoglobulins, and thromboelastography was performed. The abovementioned cells were seeded and then incubated with monoclonal antibodies to evaluate HuMab-TF cytotoxicity in vitro. The percentage of dead cells relative to positive (100% cell death) and negative (untreated cells) controls was measured. In vivo, xenograft models derived from the tumour cell line were injected into severe combined immunodeficient mice to investigate HuMab-TF.20
In vitro, the TF ADC showed high cytotoxicity against A431 and HPAF-II cells, which presented high TF levels. In contrast, the TF ADC had a limited effect in cells with low TF expression, and no activity in the negative cells, suggesting that in vitro HuMab-TF activity was dependent on TF expression.20
In vivo, the ADC containing MMAE demonstrated significant cytotoxic activity. Complete responses were observed in all mice treated with TF-011-MMAE and in most mice treated with TF-098-MMAE and TF-111-MMAE. The responses were maintained after 20–30 days following the last treatment dose, and at recurrence. No response in the presence of low TF expression was observed in xenograft models.20
Patient-derived xenografts (PDX) probably represent the best model to study heterogeneous solid tumours and are particularly helpful for evaluating the activity of TF-targeting ADCs because of non-homogeneous TF expression on the cell surface. In one study, seven PDX models were selected according to TF level of expression; in particular, in CC PDX models, TF positivity ranged from 25% to 50%. PDX models were implanted into nude mice treated with TF-011-MMAE. Surprisingly, the CC PDX models showed tumour regression despite low TF expression. This result was likely due to the capacity of MMAE to diffuse across cell membranes and act on neighbouring TF-negative cells.22
Clinical evidence
Two clinical trials have been published addressing the role of TV in CC (Table 1).23–25 The innovaTV 201 study was a phase Ib/II trial with a dose-escalation phase followed by a dose-expansion phase in patients with advanced or metastatic tumours who relapsed after or were not eligible for the standard of care.23,24 The expansion phase included patients with cervical, ovarian, endometrial, prostate, bladder, oesophageal or non-small-cell lung cancer. Eligible patients in the CC cohort had measurable disease and had received at least four prior lines of chemotherapy. Patients with coagulation defects, ongoing major bleeding or residual grade 2 neuropathy were excluded. TF expression3 (1%) was confirmed in the majority of evaluable patients (100% had membrane expression and 95% had cytoplasmatic expression); however, TF expression was not an inclusion criterion. A total of 55 patients were enrolled and received TV 2 mg/kg every 3 weeks for four cycles. In cases of stable disease, partial or complete response, patients could continue treatment up to 12 cycles and beyond if clinical benefit was still present (NCT03245736).
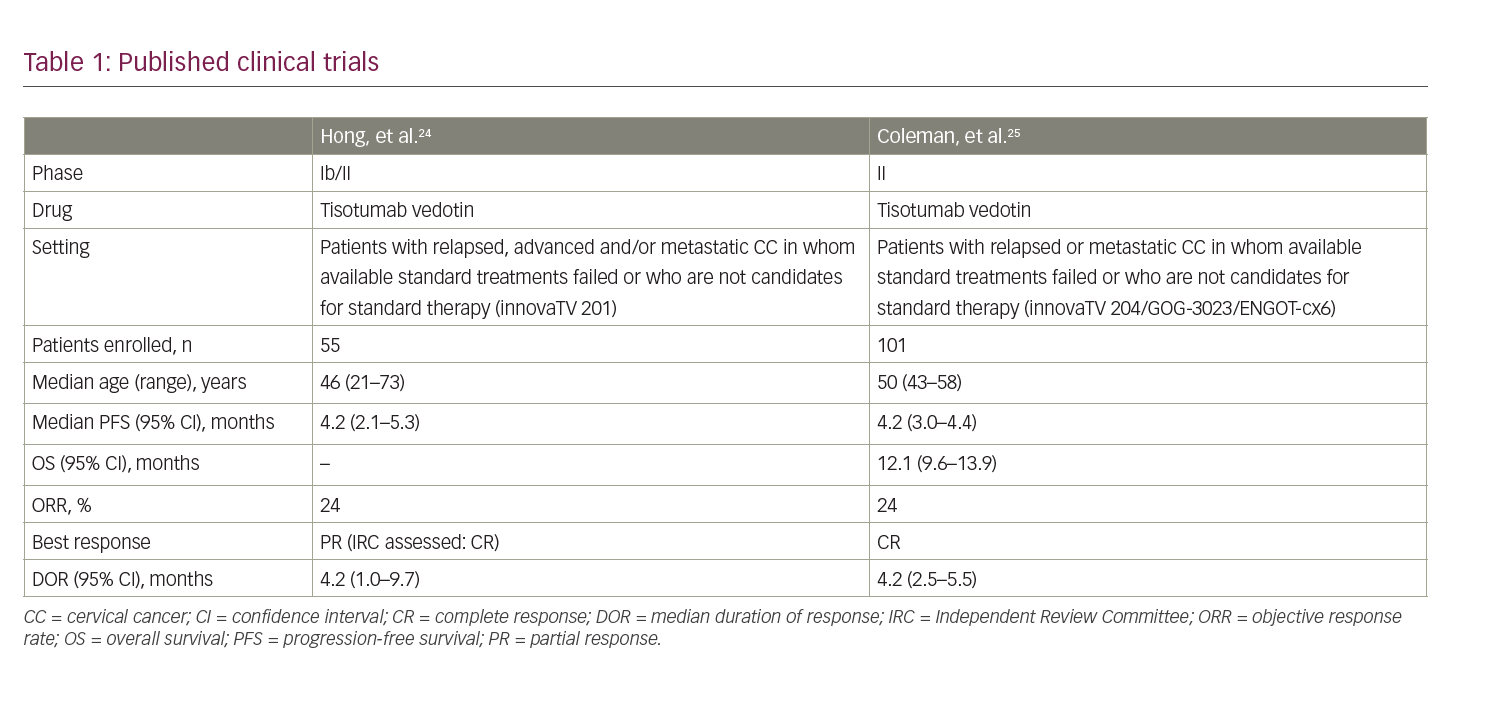
Safety was the primary endpoint. Patients received a prophylactic ocular medication with lubricating, vasoconstrictor and steroid eye drops to mitigate the risk of ocular adverse events (AEs). Overall, 56% of patients developed a grade 3 or worse AE; the most common were anaemia (11%), fatigue (9%) and vomiting (7%). All patients developed at least one AE of any grade; the most common were fatigue (51%), epistaxis (51%), nausea (49%) and conjunctivitis (42%). Among AEs of special interest (AESI), the most common were ocular events, although 10 patients (9%) discontinued treatment for peripheral neuropathy. Prophylactic medications significantly reduced the incidence of ocular events.22,23
Two fatal events occurred, both due to disease progression. Evaluation of anti-tumour activity was investigated as a secondary endpoint: the median progression-free survival (mPFS) was 4.2 months (95% CI 2.1–5.3 months); the investigator-evaluated objective response rate (ORR) was 24% (95% CI 13–37%) and was confirmed by the independent review committee (IRC) evaluation (ORR 22%).22,23
Recently, Coleman, et al. evaluated the anti-tumour activity and safety of TV administered at a dosage of 2 mg/kg every 3 weeks in patients with advanced CC who progressed after no more than two prior treatment lines.25 The primary endpoint was ORR, and secondary endpoints included safety, tolerability, PFS, OS, time to response and duration of response (DOR). The correlation between TF expression and ORR was a prespecified exploratory endpoint. Among 102 screened patients, 101 were enrolled. At a median follow-up of 10 months, four patients were still on treatment and 33 were in follow-up, with a median duration of treatment of 4.2 months and a median of 6 cycles/patient. The IRC-confirmed ORR was 24% (95% CI 16–33%) with seven complete responses (7%) and 17 partial responses (17%); disease control rate (DCR) was 72% (95% CI 63–81%) with a median time to response of 1.4 months (95% CI 1.3–1.5 months). Kaplan–Meier curves showed a mPFS of 4.2 months (95% CI 3.0–4.4 months) and a mOS of 12.1 months (95% CI 9.6–13.9 months). In the exploratory analysis, no correlation between TF membrane expression and response to TV was found.25
Safety was consistent with previously reported AEs, and treatment-related AEs occurred in 92 patients (93%). Grade 3 or worse AEs were observed in 28 patients (28%); most common were neutropenia (3%), fatigue (2%), ulcerative keratitis (2%) and peripheral neuropathies (2%). One grade 5 event was deemed related to treatment.25
According to the protocol, ocular, bleeding and neurological treatment-related AEs led to dose modification: 24% of patients had their dose interrupted, 22% had their dose reduced and 12% had their treatment discontinued. Among ocular AEs, the most common were conjunctivitis (23%), dry eye (23%) and keratitis (11%). Only two patients experienced a grade 3 AE and most of these events (86%) had completely resolved 30 days after the last dose.25
Bleeding AEs occurred in 39 patients (39%), with only two (2%) of grade 3. The most common bleeding events were epistaxis (30%), vaginal haemorrhage (7%) and haematuria (3%). More than 90% of the events had resolved by the 30-day follow-up visit.25 A total of 33 patients (33%) experienced a peripheral neuropathy treatment-related AE, with seven events (7%) of grade 3. Unlike previously reported AESIs, neuronal damage was persistent in 90 patients (79%) 30 days after the last dose of TV. Overall, 13% of patients experienced serious adverse events (SAEs), the most common of which was sensor-motor neuropathy (2%).25
Ongoing trials
Several phase I–III clinical trials are ongoing to evaluate the safety and efficacy of TV when administered alone or in combination with other drugs (Table 2).26–30 NCT0378608126 is a phase Ib/II open-label trial, investigating TV as monotherapy and in combination with bevacizumab, pembrolizumab or carboplatin in patients with recurrent or metastatic (stage IVB) CC. The trial consists of a dose-escalation phase and a dose-expansion phase. The dose-escalation phase will include patients with CC who have progressed during or after standard therapy and are ineligible for further standard treatments. This phase will be conducted to establish the maximum tolerated dose (MTD) and the recommended phase II dose (RP2D) of TV used in combination with bevacizumab (Arm A), pembrolizumab (Arm B) or carboplatin (Arm C). In the dose-expansion phase, patients who have not received prior systemic therapy will receive TV in combination with carboplatin (Arm D) or pembrolizumab (Arm E), while patients who have progressed on or after standard-of-care treatment will receive TV in combination with pembrolizumab (Arm F) or TV alone in a weekly schedule (Arm G). The primary endpoint of the expansion phase is ORR based upon the response evaluation criteria in solid tumours (RECIST) 1.1.
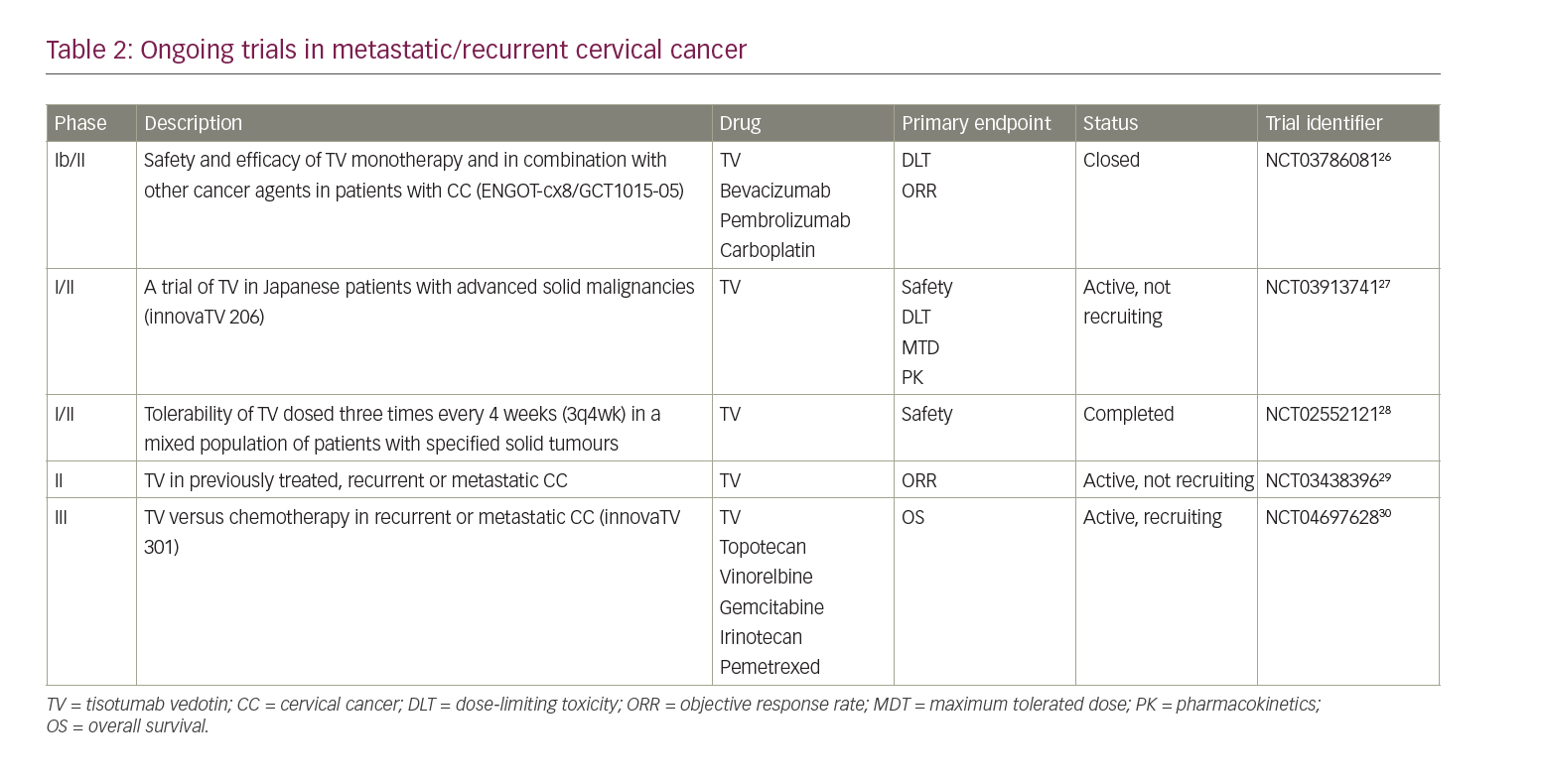
The innovaTV 206 study (NCT03913741)27 is a phase I/II, single-arm, open-label trial assessing the activity of TV in solid tumours, including CC, in Japanese patients. The study comprises two parts: part 1 aims to evaluate MTD, RP2D and the safety profile of TV in patients with several solid malignancies; part 2 will include only patients with CC who have experienced disease progression after at least two previous chemotherapy lines, and will provide data regarding safety, tolerability, pharmacokinetics and anti-tumour activity.
Another phase I/II study (NCT02552121)28 established the tolerability of TV in a mixed population of patients with solid tumours, including ovarian, cervical, endometrial, bladder, prostate (castration resistant), oesophageal and non-small-cell lung cancer. The study included a dose-escalation phase, with increasing doses of TV in 28-day treatment cycles, followed by an expansion phase to explore the RP2D of TV as determined in the first part. Safety (AEs, SAEs and infusion-related AEs) was the primary endpoint. A total of 33 patients were enrolled from November 2015 to December 2017 in two cohorts with different doses of TV (0.9 mg/kg and 1.2 mg/kg), both administered weekly three times every 4 weeks (3q4wk). Part 2 included only patients with cervical or ovarian cancer, and consisted of six cohorts designed to compare the weekly 3q4wk schedule (dose 1.2 mg/kg as defined in phase I trial) with the 3-weekly 2.0 mg/kg schedule (1q3wk). Two cohorts were added because of severe ocular toxicity experienced by some patients on the 3q4wk schedule; affected patients switched from the 3q4wk schedule to 1q3wk administration.
NCT0343839629 is a single-arm, multicentre, phase II study of TV in patients with previously treated, recurrent or metastatic CC. All patients will receive TV 2.0 mg/kg intravenously 1q3wk until disease progression or toxicity. The primary endpoint will be the ORR evaluated according to RECIST 1.1 and assessed by IRC.
The innovaTV 301 study (NCT04697628)30 is a randomized, open-label, phase III trial evaluating the efficacy of TV versus physician’s chemotherapy choice in recurrent or metastatic CC. According to the study design, 482 participants will be randomized to receive TV 2.0 mg/kg 1q3wk versus standard chemotherapy including topotecan, vinorelbine, gemcitabine or irinotecan and pemetrexed according to investigators’ choice. The primary endpoint is OS, and secondary endpoints are PFS, ORR, DOR, time to response and quality of life.
Discussion
Advanced or recurrent CC progressing on first-line platinum-based chemotherapy still represents an urgent unmet clinical need, with few effective chemotherapy agents available. Besides immunotherapy, no other promising drugs are in clinical development.
The ADC TV could represent a concrete and promising option in the second-line setting in patients who do not respond to platinum first-line treatment. The 24% ORR reported in the CX6 study25 appears particularly interesting compared with historical chemotherapy activity data; furthermore, the characteristics of the patients enrolled in this trial are very close to those of the real-world advanced CC population, where patients are mostly pretreated with bevacizumab, which seems not to affect TV efficacy or to increase its toxicity. Of note, responses were detected in both squamous and adenocarcinoma histotypes, the latter of which is apparently less sensitive to immunotherapy, and TV may therefore represent a valuable alternative for these tumours. The toxicity profile of TV in terms of ocular events and bleeding seems manageable with the support of mitigation strategies, particularly those implemented for ocular events. The management of neurotoxicity requires caution, experience and careful follow-up, as neuropathy is the only long-lasting AESI reported in early clinical trials. The combination with carboplatin, bevacizumab and pembrolizumab, if feasible and active, could represent an alternative to the standard carboplatin-paclitaxel regimen, and warrants further exploration in comparison with the standard of care.