Annually, 1.3 million cases of NSCLC occur.1 Despite smoking being the most important environmental causative factor for lung cancer, 10% of lung cancers occur in never-smokers, thus emphasizing the importance of genetic factors.2 Detection methods for genomic alterations include array-based profiling, targeted sequencing, and whole-genome sequencing. The following driver genetic alterations and respective frequencies occur in lung adenocarcinoma: EGFR (5–15%), ALK (5–15%), and KRAS (>15%). Genes with a mutation rate <5% include BRAF, PIK3CA, MAP2K1, MET, and HER2. The ability to detect such driver mutations in a majority of lung cancer patient specimens has been demonstrated in large genomic projects, such as the Lung Cancer Mutation Consortium (LCMC),3 which seeks to not only determine driver mutations but also allow clinicians to use this knowledge to use appropriate targeted therapies or enroll patients into relevant clinical trials. In 2012, next generation sequencing of 183 lung adenocarcinomas was reported.4 New findings that supplemented existing literature included recurrent somatic mutations in the splicing factor gene U2AF1 and truncating mutations in ARID1A and RBM10. Loss of function mutations and deletions in tumor suppressor genes are not easy to therapeutically target but deserve inclusion. These include LKB1, TP53, RB1, NF1, CDKN2A, SMARCA4, and KEAP1.5 Inactivation of p16Ink4 also occurs in lung adenocarcinoma and is associated with cigarette smoking. Finally, somatic focal amplifications of NKX2-1 as well as recurrent inframe fusions of KIF5B and RET can also occur.
Molecular Biology
In an assessment of 188 cases of lung adenocarcinoma in 2008, DNA sequencing was performed on 623 selected genes. Twenty-six genes emerged that were frequently mutated and considered likely involved in carcinogenesis.5 The inferred significantly mutated pathways were the MAPK, Wnt, p53 signaling, cell cycle, and mammalian target of rapamycin (mTOR) pathways. Clinically, KRAS mutations and LKB1 mutations were correlated with smoking status. EGFR mutations were correlated with never-smoker status.6 Pathologically, mutations of LRP1B, TP53, and INHBA were negatively correlated with acinar, papillary, and bronchioalveolar subtypes, but were significantly positively correlated with the solid subtype. By contrast, EGFR mutations were negatively correlated with the solid subtype and positively correlated with the papillary subtype. Molecular alterations of importance in lung adenocarcinoma are also not solely restricted to mutations, but include amplifications or gene rearrangements.
Ethnic differences exist in different studied populations with differing molecular tumor characteristics and reported frequencies. The clinician must use interpretive caution when applying data between different groups—Asian and Western populations differences are a notable example. The incidence in lung adenocarcinoma of commonly mutated genes and rearrangements in ethnically different populations is detailed in Figure 1. A study comprising an ethnically heterogeneous series of nonsmall- cell lung cancer (NSCLC) found LKB1 mutations in 17% of NSCLC of US origin compared with 5% of Korean cases (p=0.001).6 EGFR mutations are more frequent in NSCLC arising in patients of Asian ethnicity. Patterns of co-occurrence and mutual exclusivity of genomic alterations are also important. In another case series of NSCLC (n=1,683) ALK rearrangements were mutually exclusive of EGFR or KRAS mutations.7
Technical Considerations
The American College of Pathologists devised evidence-based guidelines in 2013 to select the appropriate patients and samples for EGFR- and ALK-directed therapeutics.8 The principle recommendations are to test for EGFR mutations and ALK fusions to guide patient treatment selection, in patients with lung adenocarcinoma. This directive is irrespective of gender, race, smoking history, or other clinical risk factors.
Detection methods for genetic alterations in lung adenocarcinoma include real-time polymerase chain reaction (RT-PCR), immunohistochemistry (IHC), fluorescent in situ hybridization (FISH), and next generation sequencing. The US Food and Drug Administration (FDA) has approved a FISH assay for ALK rearrangements using dual-labeled break-apart probes. IHC for ALK rearrangements has a sensitivity of 90% and specificity of 97.8 %; however, IHC for ALK rearrangements has been subject to challenge because low expression levels encoded by ALK fusion transcripts may occur.9–13 More reassuringly, 100% correlation with FISH has been more recently established using an ultrasensitive IHC technique.14 In one series IHC was concordant with FISH in 229 of 231 dual-informative samples. Difficulties with RT-PCR for ALK rearranged disease include variability in the partner gene breakpoint and partner gene identity within the gene fusion. In ROS1-rearranged NSCLC, FISH is an appropriate though expensive and technically demanding diagnostic method. RT-PCR does not detect all FISH-positive cases. The inference is that other ROS1 partners or different breakpoints to CD74-ROS1 and SLC34A2-ROS1 may emerge. Similarly, in RET fusion positive lung adenocarcinoma RT-PCR is insufficient to detect RET fusion partners or isoforms. IHC is also currently insufficiently reliable for diagnostic purposes.15–17 In terms of EGFR, the FDA have approved the therascreen EGFR RGQ (Rotor-Gene Q 5plex HRM instrument) PCR Kit to detect EGFR exon 19 deletions or exon 21 (L858R) substitutions.
Epidermal Growth Factor Receptor
Therapeutic responsiveness of NSCLC to the anilinoquinazoline inhibitors gefitinib and erlotinib is correlated with EGFR mutations. These are small deletions that affect amino acids 747–750 or point mutations, the most frequent of which is replacement of leucine by arginine at codon 858 (L858R).18,19 Other less-common mutations include G719S in exon 18 and L861Q in exon 21. EGFR mutations occur in approximately 9% of cases of NSCLC. Treatment responsiveness is clinically correlated with females, nonsmokers, adenocarcinoma histology, and East Asian ethnicity. Patient selection for treatment with EGFR tyrosine kinase inhibitors (TKIs) should be determined by genotyping rather than by clinical features. This was found in a subset analysis of the First Line IRESSA™ Versus Carboplatin/ Paclitaxel in Asia (IPASS) trial of advanced lung adenocarcinoma.20 In that study, gefitinib was found to be superior to chemotherapy with 1-year PFS rate of 24.9% versus 6.7%; hazard ratio (HR) for death or disease progression 0.74 favoring gefitinib (p<0.001). Within the EGFR mutated cohort, gefinib was more efficacious compared with chemotherapy (HR for death or disease progression 0.48), but gefitinib was ineffective in the EGFR wild type cohort (HR 2.85). 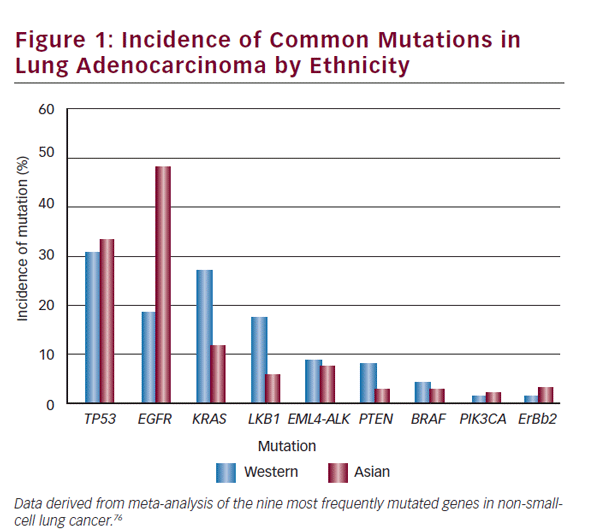
Six phase III trials have established the importance of EGFR mutation testing when considering the most appropriate treatment for newly diagnosed advanced-stage NSCLC.21 An outstanding example is an interim analysis of 200 patients enrolled onto a phase III Japanese study of gefitinib versus carboplatin-paclitaxel in advanced EGFR mutated NSCLC.22 In excess of 90% of patients in both arms had adenocarcinoma histology. PFS for all patients was 10.8 months for gefitinib versus 5.4 months for the chemotherapy-treated group; HR 0.30 (p<0.001). Of patients that progressed on chemotherapy, 95% crossed over to gefitinib and the study was prematurely terminated because of the findings of the interim analysis to the detriment of overall survival evaluation. The most-frequent toxicities from gefitinib are rash and elevated aminotransferases. Cases of gefitinibinduced severe interstitial lung disease can occur and may be fatal. This toxicity usually arises in the first 12 weeks of treatment with frequency varying by ethnicity. The frequency is 4–6% in Japan and 0.3% in the US.23,24
In 2013 the FDA approved afatinib, an irreversible EGFR/HER2 TKI, for firstline treatment of NSCLC in tumors with either an EGFR exon 19 deletions or exon 21 (L858R) substitutions. Approval was based on the findings of the international phase III trial LUX-Lung 3. In that study, 345 patients with metastatic EGFR mutant lung adenocarcinoma were assigned to either afatinib or a combination of pemetrexed and cisplatin. Median PFS was 11.1 months in the afatinib treated arm compared with 6.9 months in the chemotherapy arm (HR 0.58, 95% confidence interval [CI] 0.43–0.78; p<0.001). Within patients that had either exon 19 deletions or an exon 21 (L858R) substitution, median PFS was 13.6 months in the afatinib-treated group and 6.9 months for the chemotherapy-treated group. Also in 2013, the FDA approved erlotinib for the first-line treatment of metastatic NSCLC with an EGFR exon 19 deletion or exon 21 (L858R) substitution. This was based on the phase III Study (Tarceva®) vs Chemotherapy to Treat Advanced Non-Small Cell Lung Cancer (NSCLC) in Patients With Mutations in the TK Domain of EGFR (EURTAC ) trial. One hundred and seventy-four patients with metastatic NSCLC were treated with erlotinib or platinum-based doublet chemotherapy.25 All tumors had either an exon 19 deletion (66%) or exon 21 L858R substitution (34%). PFS was 10.4 months in the erlotinib arm and 5.2 months in the platinum-based chemotherapy arm (HR 0.34; p<0.001). 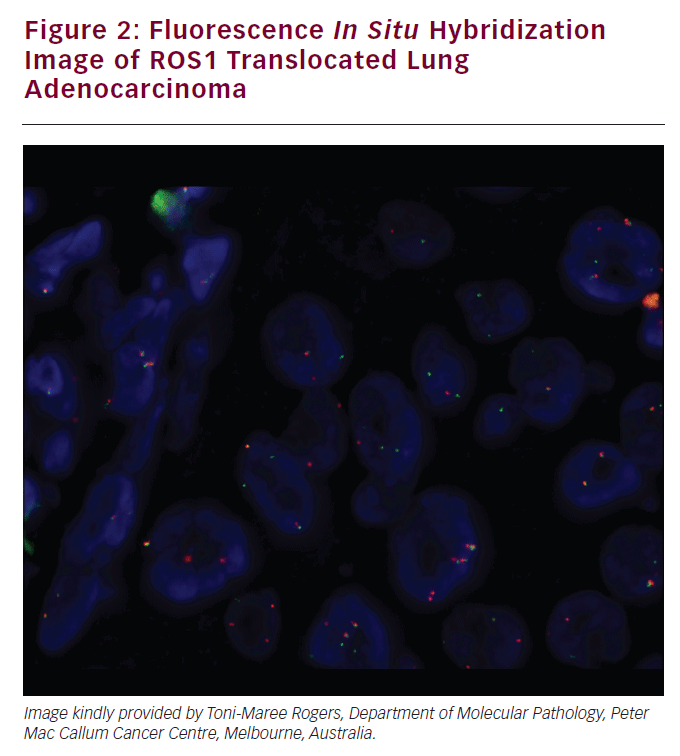
Resistance to EGFR TKI ultimately occurs usually within 1 year of TKI initiation and by different mechanisms. These include acquisition of the gatekeeper C-to-T mutation, which is predicted to change threonine to methionine (T790M).26 In a bypass phenomenon, 5–10% of cases of acquired EGFR tyrosine kinase resistance have amplification of MET, which affects downstream signaling.27 Other recently identified changes in tumors resistant to EGFR inhibitors include: PIK3CA gene mutations, EGFR amplification, epithelial-to-mesenchymal transition (EMT), and transformation to small cell histology.28
Anaplastic Lymphoma Kinase
Rearrangements of the ALK gene occur in approximately 5% of cases of NSCLC. The ALK gene encodes a tyrosine kinase receptor, which is a member of the insulin receptor superfamily. ALK is a dependence receptor and in the absence of ligand enhances apoptosis. It is physiologically important in nervous system development. ALK is located on chromosome 2p23 and ALK fusion proteins are oncogenic. The most frequent fusion partner in NSCLC is the echinoderm microtubuleassociated protein-like 4 (EML4) gene on chromosome 2p21. The EML4- ALK fusion arises by a somatic rearrangement involving inversion of a short segment of chromosome 2p. There are at least 20 variants of the EML4-ALK fusion gene alone and fusion partners other than EML4 can occur in NSCLC. While variation in the locus of the genomic breakpoint of the ALK gene fusion partner occurs, the locus of the breakpoint in the ALK gene itself is invariably constant. ALK fusion gene partners other than EML4 include TFG-ALK, KIF5B-ALK, and KLC1-ALK.29–31
Crizotinib is an oral AT P-competitive small-molecule TKI that targets ALK, MET, and ROS1.32,33 The FDA approved it for the treatment of ALK fusionpositive lung cancers in 2011.34 This was partly based on an expanded cohort study of ALK-positive cases of NSCLCs published in 2010. A preceding dose-escalation study established a recommended dose of crizotinib of 250 mg twice daily in 28-day cycles. In that study, ALK rearranged NSCLC was associated with adenocarcinoma subtype, younger patients, and those with minimal or no smoking history. The phase I study was updated in 2012. Of 143 evaluable ALK-positive patients, the objective response rate was 60.8% with mean response duration of 49.1 weeks, and median PFS of 9.7 months.35 Estimated overall survival at 12 months was 74.8%, but data remained immature. Adverse events occurred in 97% of cases including nausea, diarrhea, constipation, vomiting, visual effects, and peripheral edema. The most frequent grade 3 or 4 treatment-related adverse events were neutropenia, increased alanine aminotransferase, hypophosphatemia, and lymphopenia.
A subsequent phase III trial involving 347 patients with locally advanced or metastatic ALK-positive lung cancer was performed.36 All study entrants received one prior platinum-based regimen and were randomly assigned to crizotinib, pemetrexed, or docetaxel. Patients in the chemotherapy group were permitted to crossover to crizotinib on progression. The median PFS was 7.7 months for crizotinib compared with 3.0 months for chemotherapy (HR for progression or death in the crizotinib group was 0.49; [95% CI, 0.37 to 0.64; p<0.001]). The response rate for crizotinib was 65% compared with 20% for chemotherapy. Interim analysis found no significant improvement in survival with crizotinin compared with chemotherapy. There was, however, symptomatic improvement and superior global quality of life with crizotinib compared with chemotherapy. Crizotinib toxicities included interstitial lung disease and elevated aminotransferases. Primary or acquired resistance to crizotinib can arise by copy number aberration of the EML4-ALK fusion, ALK mutations, activation of pharmacodynamics bypass signaling, or pharmacodynamics limitations leading to the central nervous system (CNS) being a drug sanctuary site.37,38 Resistance mutations of the ALK gene include the gatekeeper mutation Leu1196Met (stearic hindrance to TKI binding) or mutations in the ALK kinase domain including Gly1202Arg, Ser1206Tyr (located in solvent-exposed region of tyrosine kinase domain), and Gly1296Ala (located in the AT Pbinding pocket).39,40 Bypass signal activation includes EGFR amplification, EGFR mutation, KRAS mutation, and amplification of Kit.39 Crizotinib has poor penetration of the blood–brain barrier with the CNS the most frequent site for disease progression. ALK inhibitors other than crizotinib are emerging with different pharmacologic properties. CH5424802 (RO5424802) is a potent selective ALK inhibitor with a spectrum of anti-tumor activity that includes Leu1196Met mutant EML4-ALK NSCLC.41 A single-arm, openlabel phase I–II study of CH542802 of patients with ALK rearranged NSCLC has occurred.42 CH5424802 was well tolerated in that trial and had high activity. A preliminary study of a second-generation ALK inhibitor LDK378 in advanced ALK-positive NSCLC has also been reported.43 LDK378 is active both for ALK rearranged NSCLC and all preclinical established crizotinibresistant mutations, including L1196M. Different doses were administered in the study with 122 patients evaluable for response. The response rate was 57% in crizotinib-resistant patients and 60% for crizotinib-naïve patients. Median response duration was 8.2 months. In particular, CNS responses associated with prolonged PFS were observed.
KRAS There are three members of the RAS family, entitled KRAS, NRAS, and HRAS. One-third of all cancers have oncogenic mutations in either codons 12, 13, or 61 of the RAS gene and 30% of adenocarcinoma have KRAS mutations. KRAS mutations can also occur in squamous cell lung tumors. KRAS mutations occur more commonly in patients who have a smoking history.44The presence of a KRAS mutation is associated with a poorer prognosis.45 There are some retrospective data to suggest that KRAS mutations may be correlated with chemotherapy resistance;46 however, KRAS mutation status has not been shown to be predictive of the benefit of adjuvant chemotherapy.47 The use of KRAS status as a predictive marker for EGFRdirected therapies has been of interest, however, when investigated in two meta-analyses this was not conclusively demonstrated to be the case.44,48
Direct therapeutic targeting of RAS has been a persistently difficult problem to overcome and remains a therapeutic lacuna.49 One strategy has been to consider MEK inhibition—a randomised, placebo-controlled phase II trial investigating the MEK inhibitor selumetinib in combination with docetaxel demonstrated a median PFS of 5.3 months in the selumetinib group and 2.1 months in the placebo group (HR 0.58, 80% CI 0.42–0.79; p=0.014).50 A recent study of NRAS mutant melanoma may suggest a possible investigational strategy for vicariously targeting KRAS mutant lung adenocarcinoma. In that study, a mouse model of NRAS mutant melanoma was leveraged and network modeling was applied. The not intuitively obvious combination of CDK4 and MEK inhibition was found to have substantial preclinical efficacy in inhibiting NRAS mutant disease using this methodology.51 An analogous tractable mouse model of KRAS-driven lung cancer and Lkb1 inactivation, discussed in the LKB1 section, may reveal proxy targets to surreptitiously inhibit KRAS mutant lung adenocarcinoma. A different preclinical strategy involving a short hairpin RNA (shRNA)-drug screen to identify combinatorial effector targets with MEK inhibition was used in a study of KRAS mutant cancers.52 The combination of BCL-XL and MEK inhibition emerged as the leading strategy with impressive in vitro and in vivo efficacy in KRAS mutant malignancy. In particular, ABT-263, which inhibits BCL-XL ability to both bind and inhibit pro-apoptotic proteins, combined with a MEK inhibitor, causes tumor regression in a KRAS-driven genetically engineered mutant mouse model of lung cancer. Emergent therapeutic strategies also include using FAK inhibitors in KRAS mutant lung adenocarcinoma. The rationale is that the RHOA-FAK network is deregulated in high-grade lung tumors. Co-expression of KRAS mutations and CDKN2A mutations is associated with aggressive treatment-resistant tumors. Suppression of RHOA/FAK selectively induced cell death in mutant KRAS:INK4A/ARF-deficient lung adenocarcinoma cells.53 Pharmacologic inhibition of FAK caused tumor regression in lung cancers, which arose in mutant Kras:Cdkn2a-null mice.
Approximately half of cases of KRAS mutant NSCLC have concurrent mutations of the tumor-suppressor gene LKB1. This accounts for approximately 7–10% of all cases of NSCLC. LKB1 tumors are not able to appropriately respond to metabolic stress, as its physiologic function normally is to activate the energy-sensing AMPK pathway. Phenformin is a former medication for type 2 diabetes and a mitochondrial inhibitor. When used as a single agent in Kras:Lkb1 compound mutant mice it prolonged survival, but was unsuccessful in Kras:p53 mice.54
LKB1
Peutz-Jeghers syndrome is caused by germ-line inactivating mutations in the serine threonine kinase LKB1 tumor-suppressor gene. Clinical features of this condition include intestinal hamartomas and epithelial cancers including NSCLC. Inactivating somatic mutations of LKB1 occur in 34% of lung adenocarcinoma and are associated with a smoking history and male gender.55,56 LKB1 mutations detected in the context of a smoking history are nearly mutually exclusive of EGFR mutations and are more frequent in Caucasians compared with Asian patients.57 In normal circumstances, upstream LKB1 directly activates and phosphorylates AMPK (AMPactivating protein kinase) as well as 12 other related kinases. AMPK itself suppresses the mTOR complex 1 (mTORC1) pathway by phosphorylating tuberous sclerosis complex 2 and raptor.58 Therefore, the effect of functional loss of LKB1 is upregulation of the mTORC1 complex pathways.
In a mutant KRAS-driven mouse model of lung cancer homozygous Lkb1 inactivation was a stronger cooperator than p53 or Ink4a/Arf.59 Inactivation of lkb1 combined with activating KRAS mutations using inducible promoters in the lung conferred decreased survival compared with KRAS mutation alone. Lkb1-deficient tumors had an expanded histologic spectrum (adenocarcinoma, squamous, and large-cell carcinoma), shorter latency, and more frequent metastasis. Expression profiling of mouse lung tumors and human lung cancer cell lines identified the metastasis-promoting genes NEDD9, VEGFC, and CD24, as targets of LKB1-repressed lung cancer. Despite LKB1 negatively regulating the mTOR pathway when A549 cells were treated with the mTOR inhibitor rapamycin, there was no effect on NEDD9 protein levels. It was found that within a subset of Kras-induced mouse tumors, loss of LKB1 expression led to transcript overexpression characteristic of human squamous cell carcinomas. Overall, it appears that LKB1 suppresses lung cancer initiation, differentiation, and metastasis.
ROS1 Rearrangements
The ROS1 tyrosine kinase gene resides on chromosome 6q22. Chromosomal rearrangements of ROS1 occur in 1.7% of NSCLC. A ROS1 translocated lung adenocarcinoma was demonstrated in Figure 2. Patients with ROS1 rearrangements NSCLC are more likely to be never-smokers and younger. In one study of NSCLC, all of the identified ROS1-positive tumors were adenocarcinomas with one-third having high-grade highly atypical infiltrating tumor cells.60 In vitro sensitivity to crizotinib was established in NSCLC cell lines and cells transfected with CD74-ROS1. A patient with ROS- 1 rearranged NSCLC was treated with crizotinib and experienced a nearcomplete response. Crizotinib is efficacious in treating lung cancers with ROS1 translocations. In one reported case, a patient with metastatic lung adenocarcinoma and a CD74-ROS1 rearrangement had an initial dramatic response to crizotinib, but developed acquired treatment resistance. Biopsy of the resistant tumor found an acquired mutation that conferred a glycine-toarginine substitution at codon 2032 within the ROS1 kinase domain. This was not a gatekeeper mutation but conferred resistance to ROS1 kinase inhibition by steric interference.61 RET RET is important in cellular proliferation, migration, neuronal navigation, and cell differentiation when it binds to the glial cell-derived neurotropic factor family of ligands.62 RET fusion occur in 1.4% of NSCLC and 1.7% of adenocarcinoma of the lung. This is increased to 6% for NSCLCs, which are pan-negative for other established driver mutations.63 A further increase to 16% occurred in a prospective phase II trial restricted to pannegative never-smokers.15 In one study of clinical–pathologic correlates RET fusion genes were assessed in a series of 936 surgically resected NSCLC.16 RET fusions were detected in 11 adenocarcinomas (n=11 of 633) and two adenosquamous cell carcinomas (n=2 or 24). Of the 13 patients, nine had a KIF5B-RET fusion, three had a CCDC6-RET, and one had an NCOA4-RET fusion. Patients with adenocarcinoma of the lung and RET fusion genes had more poorly differentiated tumors, were more likely to be younger (<60 years), and never-smokers. These tumors were also associated with a solid subtype and a smaller dimension (<3 cm) with N2 disease. Preliminary data was reported of the first three entrants onto a phase II trial of the RET inhibitor cabozantinib when used in patients with RETfusion- positive NSCLC (NCT01639508). Two of these patients had partial responses including one with a novel TRIM33-RET fusion. A third patient with a KIF5B-RET fusion had stable disease of 31 weeks. MET
MET is involved in embryology, nervous system, muscular development, and wound healing and homeostasis. It is the sole receptor for hepatocyte growth factor (HGF). The HGF receptor is the protein product of the MET gene, which resides on chromosome 7q21-q31. MET amplification is one mechanism of secondary resistance to EGFR TKIs described as kinase switch. Of tumors with secondary EGFR resistance, 20% have MET amplification.64,65 In untreated cases of NSCLC MET amplification occurs in 1.4–21% and occurs in both adenocarcinomas and squamous cell carcinomas.66–68 Binding of HGF ligand to HGF receptor causes receptor dimerization, tyrosine kinase activation, and initiation of intracellular signal transduction cascades.
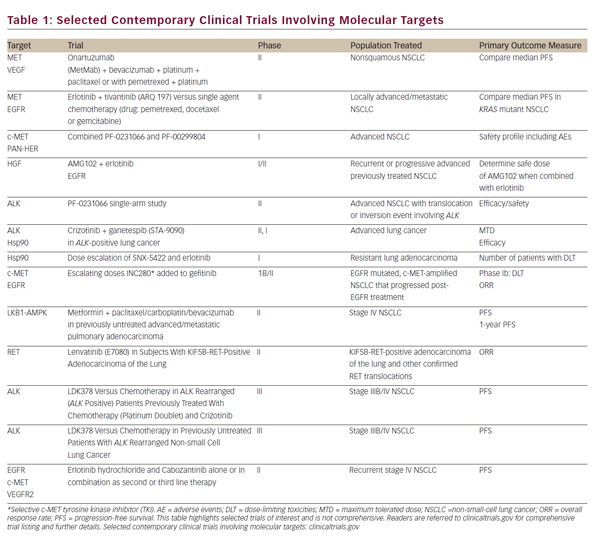
Preclinical studies have shown that MET can cause proliferation of cancer cells. In a clinical series of surgically resected NSCLC, amplification of MET was correlated with a poor prognosis.69 In one series of 188 adenocarcinomas, two somatic mutations in MET were discovered in exon 13 encoding the juxta-membrane domain (Arg988del and Tyr1021A) and one in exon 18 encoding the kinase domain (Gly1260Cys).5 Therefore, the frequency of MET gene mutations is low and furthermore their significance remains indeterminate in lung adenocarcinoma unlike other diseases, such as hereditary papillary renal cell carcinoma. In a more general context, increased MET activation other than by mutation is either by overexpression/amplification or decreased degradation. MET degradation is by E3 ubiquitin ligase c-CBL. Decreased c-CBL can arise by loss of heterozygosity and is sometime mutated in lung cancer.70 MET inhibitors are available and remain an area of ongoing research. While a number of agents have additional targets, such as vascular endothelial growth factor (VEGF) and ALK, there have also been specific MET inhibitors investigated, both mono-clonal antibodies and TKIs (outlined in Table 1). In particular, MetMab (Ornatuzumab) is a humanized modified 5D5 anti-MET monovalent antibody, which has been studied in a phase II randomized clinical trial, in combination with erlotinib—MetMab resulted in improved overall and PFS with IHC, rather than FISH, being a more sensitive predictor of benefit.71 An oral, non-AT P-dependent selective MET inhibitor, tivantinib (ARQ-197), has also demonstrated a PFS benefit in a phase II randomized clinical trial,72 in combination with erlotinib. A phase II trial is underway.
HER 2
Mutations within HER occur in a small proportion of cases of NSCLC. A study identified an in-frame insertion in exon 20 of the HER2 gene in 1.7% of 3,800 NSCLC.73 Aberrant HER2 was a driver mutation in almost all cases. All HER2 mutated tumors were adenocarcinomas 50 % of which were stage IV adenocarcinomas. This cohort was treated with HER2 directed treatments. Overall response rate was 50 % with a disease control rate of 82%.
New Advances
New promising treatment strategies emerged from the American Society of Clinical Oncology (ASCO) annual meeting in June 2013. BRAF mutations occur in 4% of NSCLC, with approximately half being V600E mutant. Most cases occur in patients with a history of smoking.74 Interim efficacy and safety data from the first 17 patients treated, on the phase II study BRF113928, was reported.75 Dabrafenib at a dose of 150 mg orally twice daily was generally well tolerated and resulted in an overall response rate of 54%. Longest duration of response was 49 weeks.
Another advance involves heat shock protein 90 (Hsp90). This is a chaperone protein important for the effect of several oncoproteins. Ganetespib is a second-generation Hsp90 inhibitor with established preclinical synergistic effects when combined with taxanes. The GALAXY 1 international phase II trial (Abstract CRA8007) enrolled 252 patients with advanced lung adenocarcinoma that had received one prior systemic treatment for NSCLC. Participants were treated on a 1:1 ratio with ganetespib + docetaxel or docetaxel monotherapy. Median PFS was 4.5 months for the combination versus 3.2 months for docetaxel alone (HR 0.83). Overall survival was 9.8 for combined treatment compared with 7.4 months for the monotherapy treatment arm (HR 0.73). Within the prespecified subgroup analysis, patients enrolled more than than 6 months postdiagnosis of advanced disease stage had a PFS of 5.4 months compared with 3.4 months (p=0.0075), favoring the combination treatment. Overall survival of 10.7 months versus 6.4 months (p=0.0036) again favored the combination treatment arm.
Conclusion
The recognition that adenocarcinoma of the lung is a molecular heterogeneous disease heralds a new era in treating this most common form of lung cancer. Subverting molecular heterogeneity and clonal evolution by sequential and combinatorial molecular therapeutic approaches will likely be the subject of research for years to come. Overall the benefit to patients is already here with further gains anticipated.