Triple-negative breast cancer (TNBC) is a biologically aggressive form of breast cancer defined by the absence of the oestrogen receptor and progesterone receptor, as well as lack of amplification of the human epidermal growth factor receptor 2 (HER2). TNBC accounts for up to 20% of breast cancers, and metastatic TNBC (mTNBC) is associated with worse outcomes compared with other breast cancer subtypes, with a median overall survival (OS) of approximately 18–19 months on chemotherapy.1–3 As TNBC is a disease defined by the absence of targetable receptors, it has been historically difficult to treat, and patients used to broadly receive only non-targeted chemotherapy.
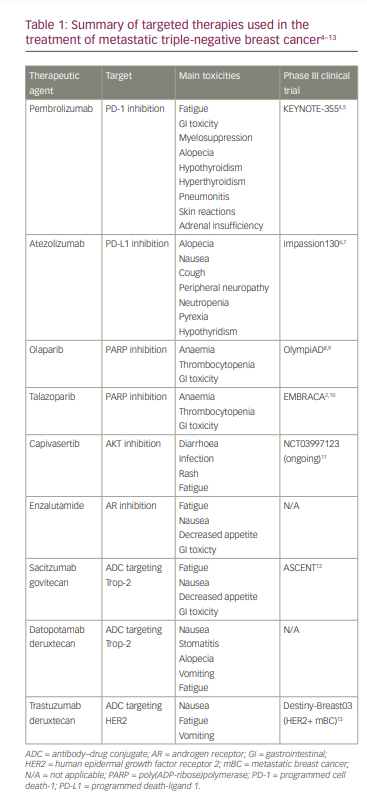
Advancing molecular diagnostic tools and corresponding clinical studies have led to further molecular characterizations of TNBC, and now therapy is informed by the presence or absence of targetable features. In this article, we review the current standards for identifying these targets and managing mTNBC using immunotherapy, poly(ADP-ribose)polymerase (PARP) inhibitors and antibody–drug conjugates (ADCs). The targeted therapies discussed are summarized in Table 1.4–13
Targeted therapies
Immunotherapy
Historically, first-line therapy for mTNBC consisted of chemotherapy without any molecular information dictating the regimen. The advent of immunotherapy and biomarker development has now transformed the first-line therapy for patients with mTNBC, particularly for patients with programmed death-ligand 1 (PD-L1)-positive disease. The interaction between programmed cell death-1 (PD-1) on T cells and PD-L1 and PD-L2 on host tissues was physiologically designed for host tissue protection against immune rejection. Cancer cells may usurp this pathway and evade tumour immune rejection by increasing expression of PD-1 on tumour-infiltrating lymphocytes and/or increasing expression of PD-L1 in cancer cells.14 One prior study demonstrated a greater presence of immune infiltration in TNBC compared with other breast cancers.15 By blocking this interaction, immunotherapy agents such as pembrolizumab (anti-PD-1 antibody) work by increasing antitumour immunity.
The phase III double-blinded KEYNOTE-355 study evaluated the addition of pembrolizumab to standard chemotherapy for mTNBC as first-line therapy.4 Of 1,372 patients screened, 847 were randomized 2:1 to receive either pembrolizumab plus chemotherapy versus chemotherapy alone. The chemotherapy partners included paclitaxel, nab-paclitaxel and gemcitabine/carboplatin. In the subset of patients with a combined positive score (CPS) ≥10, the addition of pembrolizumab resulted in a statistically significant 27% reduction in risk of death. Median OS for patients with PD-L1-positive tumours in the pembrolizumab group was 23.0 months, whereas in the chemotherapy alone group it was 16.1 months (hazard ratio [HR] 0.73, 95% confidence interval [CI] 0.55–0.95; p=0.0093). Final median progression-free survival (PFS) for PD-L1-positive tumours was 9.7 months in the pembrolizumab cohort (HR 0.66, 95% CI 7.6–11.3) compared with 5.6 months in the chemotherapy alone cohort (HR 0.75, 95% CI 5.3–7.5). The most common pembrolizumab-related toxicities included anaemia (49%), neutropenia (41%) and nausea (39%). Quality of life data revealed that the addition of pembrolizumab to chemotherapy did not result in a decrease in health-related quality of life in patients with previously untreated, PD-L1-positive mTNBC.5
The pivotal KEYNOTE-355 study results led to US Food and Drug Administration (FDA) approval of pembrolizumab as first-line therapy for patients with PD-L1-positive mTNBC in 2021. As per the clinical trial, a tumour is determined to be PD-L1 positive if the CPS is ≥10. CPS describes the percentage of PD-L1 expression on all cells within the tumour, including the tumour cells themselves, as well as any infiltrating lymphocytes or macrophages, which is much more comprehensive than assessment of PD-L1 in tumour only.
Since the KEYNOTE-355 study, pembrolizumab has also been approved by the FDA for treatment of stages II and III TNBC. This approval may impact the metastatic treatment paradigm depending on the timing of recurrences. Specifically, clinicians will need to consider whether or not to re-treat a patient with pembrolizumab in the metastatic setting if they have already received it in the early-stage setting.
Of note, atezolizumab had previously been granted accelerated approval in PD-L1-positive mTNBC in 2019 due to early results from the phase III Impassion130 study showing improved PFS (HR 0.60, 95% CI 0.48–0.77; p≥0.0001) in the atezolizumab + paclitaxel cohort compared with paclitaxel alone;6 however this approval was contingent on results of the follow-up Impassion131 trial.7 The latter failed to meet the primary endpoint of improved PFS (HR 0.82, 95% CI 0.60–1.12; p=0.20), and there was no survival advantage in either the PD-L1-positive group or the intention-to-treat group. For this reason, Genentech voluntarily withdrew atezolizumab’s breast cancer indication in 2021. Currently, pembrolizumab is the only FDA-approved therapy for patients with mTNBC.
Poly(ADP-ribose)polymerase inhibitors
Germline mutations in BRCA1 and BRCA2 (gBRCA1/2) are present in 5–10% of breast cancers,16 typically TNBC.17 Deleterious gBRCA1/2 mutations are associated with increased sensitivity to PARP inhibitors. There are multiple proposed mechanisms regarding the synthetic lethality of PARP and BRCA1/2 inhibition. The BRCA1 and BRCA2 proteins are responsible for homologous recombination repair of double-strand DNA breaks, whereas PARP1 and PARP2 are involved in repairing single-strand DNA breaks.18 When PARP is inhibited, single-strand breaks are converted to double-strand breaks, which require homologous recombination in order to be repaired. If BRCA1/2 is mutated, cells cannot undergo homologous recombination, and the result is cell death. PARP inhibitors may also become trapped at the DNA level, which prevents DNA replication.19 Accordingly, in patients with deficient BRCA1/2, the inhibition of PARP leads to synthetic lethality.
In the phase III OlympiAD trial,8 302 patients with gBRCA1/2 mutations were randomized 2:1 to either olaparib, a PARP inhibitor, or standard single-agent chemotherapy (capecitabine, eribulin or vinorelbine). Median PFS was significantly longer in the olaparib group than in the standard therapy group (7.0 months versus 4.2 months; HR 0.58, 95% CI 0.43–0.80; p<0.001). Notable adverse events in patients receiving olaparib included anaemia, vomiting, fatigue, headache and cough. Health-related quality of life was consistently improved for patients treated with olaparib compared with patients treated with chemotherapy.9 The phase III EMBRACA study2 randomized 431 patients with gBRCA1/2 mutations in a 2:1 ratio to receive talazoparib, another PARP inhibitor, or standard single-agent therapy (capecitabine, eribulin, gemcitabine or vinorelbine). Patients receiving talazoparib experienced a median PFS of 8.6 months compared with 5.6 months in the standard therapy group (HR 0.54, 95% CI 2.9–8.8; p<0.001). Common toxicities associated with talazoparib included anaemia, fatigue and nausea. Patients who received talazoparib had significant overall improvements in quality of life and breast cancer-specific symptoms.10
The pivotal OlympiAD and EMBRACA trials led to the FDA approval of olaparib and talazoparib, respectively, for pretreated metastatic gBRCA1/2, HER2-negative breast cancer in 2018. When a patient presents with PD-L1-positive mTNBC with a gBRCA1/2 mutation, the preference is generally to consider immunotherapy first because there is a survival advantage with immunotherapy and immunotherapy tends to work better in earlier lines. However, the order in which PARP inhibitors and pembrolizumab should be given in cases such as this has not been explicitly evaluated in clinical trials and further research is needed.
Other targeted therapies
Approximately 45% of breast tumours may harbour mutations in the AKT pathway, including PIK3CA, AKT1 and PTEN.20,21 These observations incited interest in AKT inhibitors for TNBC. The phase II double-blind, placebo-controlled PAKT trial22 investigated capivasertib, an AKT inhibitor, with paclitaxel versus paclitaxel alone as first-line treatment of mTNBC. In patients with PIK3CA/AKT1/PTEN alterations who were randomized to the investigational arm, PFS improved significantly (9.3 versus 3.7 months; HR 0.30; p=0.01). A phase III study is currently ongoing in patients with advanced TNBC (NCT03997123).11
A subset of TNBC expresses androgen receptor (AR).23 One AR inhibitor, enzalutamide, was evaluated in a phase II study conducted in advanced AR-positive TNBC.24 AR positivity was defined by immunohistochemical (IHC) staining >0%. Median PFS was 3.3 months (95% CI 1.9–4.1) in patients with one or more postbaseline assessment whose tumour expressed ≥10% nuclear AR, and 2.9 months (95% CI 1.9–3.7) in the intention-to-treat population. Of note, there is a lack of consensus on the best way to define AR positivity, given significant variability within IHC and genomic assays, and within studies that have been conducted using them.
AKT inhibitors and AR inhibitors are not currently FDA approved. We eagerly await the results from the empirical clinical trials.
Antibody–drug conjugates
Second- and third-line therapies for mTNBC have been improved by the advent of ADCs, which selectively deliver cytotoxic agents to cancer cells through coupling with monoclonal antibodies. In the basic construct of an ADC, the antibodies bind to receptors on cancer cells and then are incorporated into the cell via receptor-mediated endocytosis. The ADC is then processed in the lysosome, where the linker is cleaved, and the chemotherapeutic payload is released. The payload is a potent chemotherapeutic agent, and because it is selectively delivered to cancer cells instead of systemically administered, the potency can be 100 to 1000 times more concentrated than systemic chemotherapy.25
Sacitzumab govitecan-hziy (SG) is one ADC that combines an irinotecan metabolite (SN-38) to an anti Trop-2 monoclonal antibody with a cleavable linker.26,27 Trop-2 is a transmembrane glycoprotein that is widely expressed in most solid tumours (such as breast cancers), including TNBC, and promotes tumour growth and invasion.28 For these reasons, Trop-2 is a new molecular target in the treatment of mTNBC. SG has a high drug-to-antibody ratio (7.6:1) and, unlike the basic ADC construct, does not require internalization and enzymatic cleavage by the tumour cell for the liberation of SN-38 from the antibody. The linker between the antibody and SN-38 is only moderately stable, thus it can be cleaved in the acidic microenvironment of the tumour prior to internalization. This allows for hydrolysis of the linker and the subsequent release of SN-38 into the tumour microenvironment, providing a bystander effect. Thus, even cells not directly bound by the ADC may receive the therapy.
In April 2020, the FDA granted accelerated approval for SG in mTNBC based on the impressive results from a single-arm phase II multicenter trial conducted in heavily pre-treated patients (median of 3 previous therapies).29 Of 108 patients, three had complete responses and 33 had partial responses; the response rate was 33.3% (95% CI 24.6–43.1). The median PFS was 5.5 months (95% CI 4.1–6.3), and median OS was 13.0 months (95% CI 11.2–13.7). The most common grade 3 and 4 adverse events were anaemia and neutropenia, with 10 patients experiencing febrile neutropenia.
The confirmatory approval of SG was contingent on the results of the randomized phase III ASCENT study, in which it was compared with treatment of physician’s choice (eribulin, vinolebrine, capecitabine or gemcitabine) in patients with previously treated mTNBC.12 A total of 529 patients (61 with brain metastases) were randomized 1:1 to receive SG or chemotherapy. The median PFS was 5.6 months with SG (HR 0.41, 95% CI 4.3–6.3; p<0.001) and 1.7 months with chemotherapy (HR 0.41, 95% CI 1.5–2.6; p<0.001). The median OS was 12.1 months with SG (HR 0.48, 95% CI 10.7–14.0; p<0.001) and 6.7 months with chemotherapy (HR 0.48, 95% CI 5.8–7.7; p<0.002). Objective response rate was also markedly higher with SG, at 35% compared with 5% with chemotherapy. Key grade 3 or higher treatment-related adverse events were neutropenia (51% with SG and 33% with chemotherapy), leukopenia (10% and 5%), diarrhoea (10% and <1%), anaemia (8% and 5%) and febrile neutropenia (6% and 2%). As Trop-2 is broadly expressed in TNBC cells, molecular testing is not required for SG use. In April 2021, full approval in the second line (and plus) setting was granted to SG, the first ADC fully approved for treatment of patients with mTNBC.
Upcoming therapies
Following the success of SG in the triple-negative setting, other novel ADCs are in advanced stages of study, including datopotamab deruxtecan (Dato-DXd), a differentiated Trop-2-directed ADC with three components: a humanized anti-Trop-2 immunoglobulin G1 monoclonal antibody, a topoisomerase 1 inhibitor payload and a tetrapeptide-based cleavable linker. Dato-DXd is currently being tested in a phase III study in advanced hormone receptor-positive breast cancer, as well as in the phase I TROPION-PanTumor01 trial in TNBC (NCT03401385),30 among other solid tumours.31 Preliminary results of the latter have shown promising antitumour activity with a manageable safety profile in heavily pretreated patients with mTNBC.32 As of the data cutoff, 43 patients had been treated with Dato-DXd, 27 of whom remained on treatment and 16 discontinued due to disease progression. Among 38 patients evaluable for response, the overall response rate was 39% (15 partial responses, with 12 confirmed and three pending confirmation). The disease control rate was 84% (32/38). The most common treatment-related toxicities were nausea (58%), stomatitis (53%), alopecia (35%), vomiting (35%) and fatigue (33%).
In addition, recent insights have suggested that HER2-low status may represent a new actionable target for treating mTNBC. HER2 status in breast cancer exists on a threshold because it is determined based on the extent of the receptor’s amplification in tumour cells, rather than its complete presence or absence. For this reason, ADCs developed for the treatment of HER2-positive breast cancer (defined by IHC 3+ score or IHC 2+ and positive in situ hybridization [ISH] score) may also be effective in treating HER2-low breast cancer (defined by IHC 1+ or IHC 2+ and negative ISH), particularly ADCs with bystander effect. One such ADC, trastuzumab deruxtecan (T-DXd), consists of a humanized anti-HER2 monoclonal antibody linked to a topoisomerase I inhibitor payload through a tetrapeptide-based cleavable linker.33 It is uniquely designed with a high drug-to-antibody ratio of approximately 8 that remains stable, thereby delivering a potent cytotoxic payload that is internalized and selectively cleaved by lysosomal enzymes that are overexpressed in cancer cells.34 The efficacy of T-DXd in HER2-low breast cancers is attributed to the membrane permeability of its cytotoxic payload, which causes a bystander effect.13 Through this mechanism, the highly potent payload is released specifically within the HER2-overexpressing tumour cell to induce DNA damage, thereby inciting apoptosis of both the target tumour cells and the neighbouring tumour cells.
The phase III Destiny-Breast03 study randomized 524 patients with HER2-positive metastatic breast cancer (mBC) 1:1 to receive T-DXd or trastuzumab emtansine (T-DM1).13 At the first reported interim analysis, 75.8% of patients in the T-DXd arm were alive without disease progression at 12 months (95% CI 69.8–80.7) compared with 34.1% of patients in the TDM1 cohort (HR 0.28, 95% CI 0.22–0.37; p<0.001). The most common drug-related adverse events in the T-DXd arm were nausea (72.8%), fatigue (44.7%) and vomiting (44.0%).
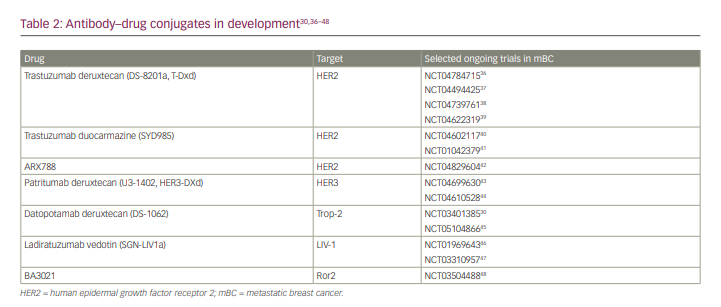
Due to the success of T-DXd in treating HER2-positive mBC, studies are now being conducted to evaluate its benefit in patients with HER2-low mBC. One such study is the phase III Destiny-Breast04 trial. The study recently reported statistically significant and clinically meaningful improvement in both PFS and OS in patients with HER2-low mBC, including mTNBC.35 No new safety concerns were identified. These results are a step forward in treating the broad spectrum of HER2 expression, and specifically present a new actionable target in breast cancer previously considered to be HER2-negative. A detailed discussion of HER2-low breast cancer is outside the scope of this article and is ideally the subject of a future review article. A summary of the different ADCs in clinical development for patients with breast cancer is provided in Table 2.30,36–48
Conclusion
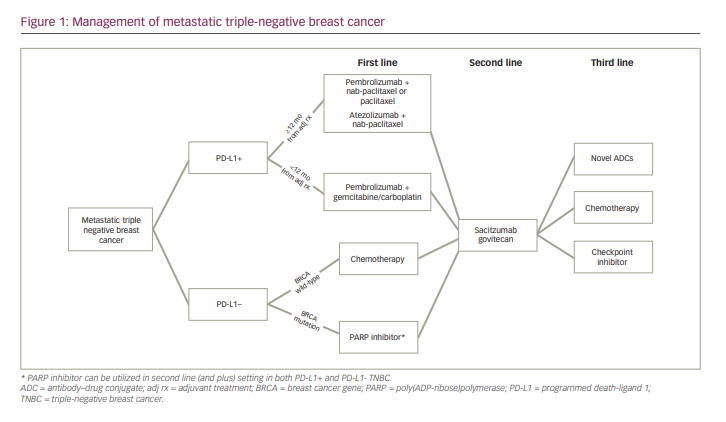
In summary, current standard of care now requires mTNBC to be evaluated for the presence or absence of PD-L1, and all patients with mTNBC should be evaluated for germline mutations in BRCA1 and BRCA2. Patients with PD-L1-positive breast cancer receive pembrolizumab in addition to chemotherapy in the first line, and patients with germline BRCA1/2 mutations may receive monotherapy with one of two PARP inhibitors, olaparib or talazoparib. In the second line and beyond, SG is the preferred treatment, consistent with FDA label and National Comprehensive Cancer Network guidelines. Later-line therapy in mTNBC could include other chemotherapy agents such as eribulin, navelbine and others. There are several novel drugs in clinical development, and clinical trial participation is always encouraged. The current treatment algorithm for management with mTNBC, as determined by the authors, is summarized in Figure 1. While TNBC is a heterogeneous disease, research has revealed various actionable targets, allowing TNBC to be subtyped in ways that were previously not possible.