Knowledge regarding the mechanisms driving progression of prostate cancer (PC) has improved dramatically in the past few years. This has allowed the accelerated development and approval of multiple drugs with different targets. The wide spectrum of rapid developments in this field warrants a comprehensive review. In this paper we aim to summarize the current standard of care incorporating recent advances, and placing emphasis on novel developments.
PC is the second most common malignancy in American men, and the second leading cause of cancer-related death in men in the US. It is estimated that in 2014 nearly 233,000 men will be diagnosed with PC, and 29,480 will die from the disease.1,2 The majority of them are diagnosed as a result of screening, so symptomatic presentation is unusual. Median age at diagnosis is 66 years. In the Surveillance Epidemiology End Results (SEER) 18 data, only 4 % have distant disease at presentation and the 5-year survival rate is 25–30 %.3
Over 95 % of PCs are adenocarcinomas. With the advent of multiple therapies, the incidence of variant histologies such as neuroendocrine PC appears to be increasing. The therapeutic management of these histologies is an area of active clinical and research interest. The therapies chosen in every setting of PC should demonstrate improved survival, but also factor in the patient characteristics of a predominantly elderly population with numerous comorbidities and the impact on quality of life (QoL).
Androgen-sensitive Metastatic Prostate Cancer (see Figure 1)
PC is predominantly driven by androgen-induced activation of androgen receptors (ARs) for its growth and survival. Disruption of this pathway ceases PC proliferation and induces apoptosis. The role of androgen ablation or androgen deprivation therapy (ADT) to suppress the testosterone to castrate levels, a Nobel Prize-winning discovery, was clearly established decades ago and continues to be the standard of care. Recent data (in the era of prostate-specific antigen [PSA] use) indicate that the current median survival for this group of patients is 5 years, but contemporary overall survival (OS) is likely to be longer given the multiple therapies that are now available for clinical use.4,5
The question of whether intermittent ADT should be the standard was put to rest with the results of Southwest Oncology Group (SWOG) 9346 (INT-0162). This was an intergroup phase III noninferiority trial in which men with androgen-sensitive metastatic disease were randomized to either intermittent or continuous ADT, if they achieved a PSA of 4 or below after 7 months of ADT. Over 3,000 patients were registered, and 1,535 patients were randomized. Median survival was 5.8 years in the continuous arm and 5.1 years in the intermittent arm (hazard ratio [HR] 1.09; 95 % confidence interval [CI] 0.95–1.24). This difference did not meet prespecified criteria for noninferiority for the study population. QoL analyses demonstrated modest but short-lived improvements in sexual function with the intermittent therapy. Based on these results continuous ADT remains the standard of care, based on optimal survival outcomes, but careful consideration of individual patient risks and counseling regarding these study results is advised.6
Gonadotropin-releasing hormone (GnRH) antagonists were developed, with the theoretical advantage of avoiding flare phenomenon and lack of testosterone surge with repeated administration. In a phase III prospective trial, two doses of GnRH antagonist degarelix were compared with leuprolide. In this study, PC patients of any stage were included. Therapy was maintained for a year. The primary endpoint of suppressed testosterone levels to ≤0.5 ng/ml at all monthly measurements was achieved in 97 % of patients in the two degarelix dose groups, which is comparable to 96 % of leuprolide-treated patients. On day 3, the median testosterone levels was reduced by >90 % in the degarelix-treated groups compared with a 65 % surge in the leuprolide group. The median PSA levels at 14 and 28 days were significantly lower in the degarelix groups than in the leuprolide group (p<0.001). The hormonal side-effect profiles were similar to previously reported effects for ADT. The injection site reactions were higher in the degarelix arm (40 % versus <1 %; p<0.001). This study confirmed that degarelix induced rapid suppression of testosterone and PSA levels with no testosterone flares.7
Combined androgen blockade (CAB) was explored to see if AR blockade adds any survival advantage. A meta-analysis was conducted of 21 randomized clinical trials comparing single-agent GnRH agonist with CAB. The study concluded that at 2 years, there was no added benefit with CAB (HR 0.970, 95 % CI 0.866–1.087) compared with monotherapy, but at 5 years there was a modest and statistically significant survival advantage with CAB (HR 0.871; 95 % CI 0.805–0.942).8 CAB remains the current standard; however, given the small incremental benefit the addition of anti-androgen (AA) can be avoided if necessary. Despite the initial response to ADT, all patients will eventually develop castrate resistance within a median timespan of 2–3 years.9
Early chemotherapy in hormone-sensitive PC has been explored as a means to delay castration resistance and to reduce morbidity and mortality. In Groupe d’Etude des Tumeurs Uro-Génitales-French Association of Urology (GETUG-AFU) 15/0403, a phase III open label trial, men with metastatic hormone-sensitive PC were randomized to ADT alone or ADT with docetaxel for up to nine cycles. The addition of chemotherapy increased the time to clinical progression by 8 months (23.46 versus 15.44 months, HR 0.75, 95 % CI 0.59–0.94; p=0.015). However there was no significant difference in the median OS, the primary endpoint of the study (58.9 months with docetaxel + ADT versus 54.2 months with ADT alone).10
The role of early chemotherapy with docetaxel in addition to ADT was evaluated the Eastern Cooperative Oncology Group-3805 (ECOG-3805) ChemoHormonal Therapy versus Androgen Ablation Randomized Trial for Extensive Disease in Prostate Cancer (CHAARTED) trial. In this phase III study, 790 men with metastatic hormone-sensitive PC were randomized to receive the combination versus ADT. A maximum of six cycles of docetaxel chemotherapy were administered. Patients were stratified by the extent of metastatic disease as high- versus low-volume. High volume was defined as the presence of visceral metastases and/or four or more sites of bone metastases. The interim results were presented at the American Society of Clinical Oncology (ASCO) 2014 annual meeting. Two-thirds of patients in each arm were classified as having high-volume disease and threequarters of men in each arm had received no prior localized treatment. As of January 2014, with a median follow-up of 29 months, there were 101 deaths in the combination arm compared with 136 deaths in the ADT arm. Median OS was 57.6 months in the combination arm and 44.0 months in the ADT arm (HR 0.61, 95 % CI 0.47, 0.80; p=0.0003). In men with highvolume disease, median OS was improved by 17 months with the addition of chemotherapy (49.2 months versus 32.2 months, HR 0.60, 95 % CI 0.45, 0.81; p=0.0006). In men with low-volume disease, median OS had not been reached at the time of this analysis and follow-up is ongoing. This study helped prove the value of early addition of chemotherapy to ADT. The toxicity profile was reasonable with 6 % of men in the chemo-hormonal regimen experiencing febrile neutropenia.
Novel agents in terms of evaluation include an insulin-like growth factor 1 receptor (IGF-1R) antibody cixutumumab (IMC-A12), which was evaluated in the SWOG 0925 trial, a randomized phase II study that has recently completed accrual (NCT01120236). The preliminary results of this study indicate lack of benefit with the addition of cixutumumab. SWOG 1216 is a planned intergroup phase III trial randomizing men to either ADT and bicaluatmide or to ADT and orteronel (TAK700), a CYP 17A inhibitor with selectivity for CYP 17, 20 lyase. Ongoing studies are evaluating the impact of early addition of androgen pathway targeted agents, such as enzalutamide, in advanced hormone sensitive disease. Randomized trials evaluating the addition of docetaxel in the PSA progression nonmetastatic disease setting have completed accrual and the results are awaited.
Despite a high and, in many cases, durable response to ADT, most patients will eventually progress to castration resistance. This remains a terminal and incurable condition; however, recently, major strides have occurred in the therapeutics of this disease state.11–13
Metastatic Castrate-resistant Prostate Cancer
Metastatic castrate-resistant PC (mCRPC) is defined as a rise in serum PSA level and/or radiographic evidence of disease progression despite castrate levels of testosterone. mCRPC is a heterogeneous disease that is marked by an array of genetic and epigenetic lesions mirrored by a spectrum of phenotypic presentations. Treatment modalities available in this setting include hormonal agents, chemotherapy, vaccines, new targets, radiation treatments, etc.14 An emerging clinical factor in these advanced, late-stage diseases is that about 25 % have neuroendocrine differentiation. They mainly present with visceral metastases, are non-PSA secretory, and are unresponsive to androgen ablation. This phenotype is seen de novo at diagnosis or can be unmasked/induced in reaction to selective treatment pressure following AR signaling therapies.
Hormonal Manipulation Therapies
The current standard of care is to continue the gonadal androgen suppression with luteinizing hormone-releasing hormone (LHRH) agonists/antagonists.
There is no clear consensus on the sequencing of secondary hormone therapies (SHM) as this has been primarily dictated by patient preference and drug availability. The SHM of historical interest include diethylstilbestrol (DES) and megestrol acetate. DES was abandoned secondary to thromboembolic and cardiovascular complications. Megestrol acetate caused low response rates of 10–15 % and was also dropped for cardiovascular toxicity. In a small retrospective chart review low-dose DES of 1 mg daily is noted to have caused cause PSA responses in about two-thirds of the patients with a median time to progression (TTP) of 16.4 weeks. The main side effect experienced was gynecomastia (59 %) and the risk for thromboembolic events was <5 % in this series.15 This therapy is suboptimal and toxic compared with contemporary therapies.
Addition of AAs is considered when chemical disease progression is noted on ADT monotherapy. A nonsteroidal AA can be considered, with a nondurable response rate of 15–20 % and no clear survival benefit. Bicalutamide 50 mg oral dose daily is felt to be most convenient with the best toxicity profile in this class.16
AA withdrawal should routinely be conducted at progression. Occasional responses are noted with median response duration of 4–6 months.17 The next reasonable agent is low-dose corticosteroids. In recent studies, prednisone 5 mg twice daily, is noted to cause PSA and objective responses of 24 % and 16 %, respectively, with PSA progression-free survival (PFS) of about 6 months.18 Other agents that are frequently used include ketoconazole, an antifungal agent that it inhibits both CYP 17 and CYP 3A4 in both gonads and adrenal glands. Randomized data with ketoconazole 400 mg three times a day (TID) along with hydrocortisone and AAWD showed a 20 % objective response rate, nondifferent from low-dose steroids.19 It is also noted that low-dose ketoconazole (200 mg TID) elicits similar PSA responses as a standard dose.20
Another antifungal agent, itraconazole, is noted to delay tumor growth by its anti-angiogenic and anti-Hedgehog properties in murine xenograft models. This was studied in a noncomparative randomized phase II design, and high-dose itraconazole (600 mg/day) is noted to have a modest antitumor activity with PSA PFS at 6 months of 48 % and above all, interestingly, this is not mediated by testosterone suppression.21
Novel Therapies Affecting the Androgen Receptor Pathway
Increasingly potent suppression of AR signaling is a prime therapeutic target in patients with mCRPC. The first proof-of-principle agent in this category was abiraterone.
Abiraterone acetate is an oral irreversible androgen biosynthesis inhibitor, with selective dual inhibition of 17 alpha hydroxylase and C 17, 20-lyase with decreased gonadal and extragonadal androgen synthesis. In the Cougar Abiraterone Acetate (COU-AA)-301 trial, abiraterone acetate (1,000 mg once daily) plus prednisone was compared with placebo plus prednisone in patients who received prior docetaxel. Statistically significant differences in OS (median 15.8 versus 11.2 months, HR 0.74; 95 % CI 0.64 to 0.86; p=0.001), time to PSA progression (median 8.5 versus 6.6 months, HR 0.63, 95 % CI, 0.58 to 0.78; p<0.001), radiographic PFS (rPFS) (median 5.6 versus 3.6 months, HR 0.66, 95 % CI 0.58 to 0.76; p<0.001), and PSA response (29.5 % versus 5.5 %; p<0.001) were detected. All endpoints were in favor of abiraterone acetate compared with prednisone plus placebo. Median time to functional decline was longer with abiraterone acetate at 5 versus 3 months (p<0.001), and significant improvements in patient-reported fatigue were noted.22–25
Given these exciting results, abiraterone was further investigated in the predocetaxel setting in COU-AA-302, where asymptomatic or minimally symptomatic were randomized in a double-blind, placebo-controlled trial. A statistically significant PFS improvement was detected with abiraterone therapy (HR 0.43, 95 % CI 0.35 to 0.52; p<0.001). A significant benefit in secondary outcomes—including time to opiate use, time to pain progression, chemotherapy initiation, functional status deterioration, and PSA progression—were in favor of abiraterone acetate (all p<0.01). The updated OS data were reported at the European Society for Medical Oncology (ESMO) meeting in September 2014 and median OS was 35 months with abiraterone and 30 months with placebo (HR 0.79, 95 % CI 0.66–0.95; p=0.0151). The rPFS was significantly prolonged in the abiraterone arm, 16.5 months, compared with the placebo arm 8.2 months (HR 0.52, 95 % CI 0.45–0.61; p<0.0001). This trial has confirmed that abiraterone delays disease progression, pain, and functional deterioration with a favorable safety profile.26
Given these promising and encouraging results, the Food and Drug Administration (FDA) has approved abiraterone acetate in both chemonaïve and postchemotherapy settings. A small percentage of patients in the above two trials have exhibited primary abiraterone resistance. This has provoked to identify PC biomarkers that may predict abiraterone sensitivity. One potential biomarker identified is TMPRSS2-ERG. It is a fusion gene of androgen-dependent growth factor and transcription factor. It is present in about 50 % of newly diagnosed PCs. In a study of 77 patients treated with abiraterone, 80 % of patients who have experienced a >90 % decline in PSA levels were TMPRSS2-ERG fusion positive.27 This was further analyzed in the COU-AA-302 study population, and a trend towards superior PFS in abiraterone-treated patients with this ERG rearrangements in tumor specimens was noted (22 versus 16 months, HR 0.59, 95 % CI 0.30–1.16; p=0.12), but not with placebo.28 Based on the finding that poly (ADP ribose) polymerase-1 (PARP-1) may be required for the survival and progression of the malignant phenotype in ERG-positive cells,29 a phase II trial adding a PARP-inhibitor, veliparib, to abiraterone is currently recruiting men with mCRPC. Patients will be stratified for the presence of the TMPRSS2-ERG fusion and randomized either to abiraterone plus veliparib or abiraterone alone (NCT01576172).
The other viable strategy explored is inhibition of AR signaling. Enzalutamide was developed for the same reason and is known to have triple action of competitive inhibition of AR binding, AR nuclear translocation, and interaction with DNA. In contrast to bicalutamide it does not have agonist properties. The Safety and Efficacy Study of MDV3100 in Patients With Castration-Resistant Prostate Cancer Who Have Been Previously Treated With Docetaxel-based Chemotherapy (AFFIRM) trial evaluated enzalutamide in docetaxel-refractory mCRPC. At a planned interim analysis, the estimated median survival was 18.4 months compared with 13.6 months in the placebo arm (p<0.0001). A 37 % reduction in the risk for death was noted. There is also a significant improvement in other endpoints: rPFS, PSA response, time-to-first skeletal-related event, and QoL.30 Subsequently, the Safety and Efficacy Study of MDV3100 in Patients With Castration-Resistant Prostate Cancer Who Have Been Previously Treated With Docetaxel-based Chemotherapy (PREVAIL) study evaluated its role in chemo-naïve mCRPC population; 1,717 patients were randomly assigned to either enzalutamide (160 mg) or placebo once daily. The first interim analysis demonstrated an OS advantage with enzalutamide over placebo (median 32.4 versus 30.2 months, HR 0.70, 95 % CI 0.59–0.83; p<0.0001). In addition, there was a significant improvement in PFS with enzalutamide compared with placebo (HR 0.19, 95 % CI 0.15–0.23; p<0.0001). As a result, the data and safety monitoring committee recommended stopping the trial and crossing the placebo patients to enzalutamide. Fatigue and hypertension were the most common clinically relevant adverse events associated with this treatment.31,32 Enzalutamide received FDA approval in both postdocetaxel and, recently in October 2014, chemo-naïve mCRPC patients.
Although these newer agents, enzalutamide and abiraterone, are breakthroughs in the treatment of mCRPC approximately 20 to 40 % of patients have primary resistance with no PSA responses. Those with initial response can eventually acquire secondary resistance. This could be plausibly explained by the presence of AR splice variants. These alternatively spliced variants encode a truncated AR protein that lacks the C-terminal ligand-binding domain but retainsthe transactivating N-terminal domain. The resultant product is constitutively active and is independent of the ligand. Although multiple AR variants are noted, AR-splice variant 7 (AR-V7) is the only variant with a functional protein detected in clinical specimens. Antonarakis et al. prospectively evaluated the presence of AR-V7 messenger RNA (mRNA) in circulating tumor cells (CTC) of patients receiving either enzalutamide or abiraterone and the outcomes. In this study, of the 31 enzalutamide-treated and 31 abiraterone-treated patients, 39 % and 19 % were noted to have a detectable AR-V7 in CTC, respectively. In both the treatment groups, AR-V7 positive patients had lower PSA response rates, shorter PSA PFS, clinical or rPFS, and OS.
The association between AR-V7 detection and therapeutic resistance was maintained after adjustment for expression of full-length AR mRNA. These results needs to be validated prospectively and in a larger sample size trial.33
There are several new promising hormonal agents in development. Orteronel (TAK-700) is an oral, nonsteroidal 17, 20-lyase inhibitor with high specificity for its target. A phase I/II dose escalation trial showed that it has lowered the testosterone levels profoundly to <1 ng/dl in the majority of patients, and in the phase II portion about half of them noticed a >50 % drop in PSA levels.34 In this study it was studied without steroids.
In the phase III study, orteronel is studied with prednisone in the postdocetaxel setting and was terminated as it failed to meet its primary endpoint OS benefit. The median OS was 17.0 months (95 % CI 15.2, 19.9) in orteronel versus 15.2 months (95 % CI 13.5, 16.9) in placebo (HR 0.886, 95 % CI 0.739, 1.062; p=0.1898).35 In the Evaluation of the Lyase Inhibitor Orteronel in Metastatic Prostate Cancer 4 (ELM-PC4) trial this was evaluated in the chemo-naïve mCRPC population. It showed a significant improvement in rPFS (median 13.8 versus 8.7 months, HR 0.7, 95 % CI 0.6–0.8; p<0.00001), but no statistically significant improvement in OS (31.4 versus 29.5 months, HR 0.9, 95 % CI 0.8–1.1; p=0.314) was noted.36
Galeterone (TOK-001) is an oral steroidal CYP 17 inhibitor, with AR antagonism and is also noted to decrease intratumoral AR levels. In the ARMOR1 phase I trial, 22 % of patients demonstrated a >50 % decrease in PSA with an overall mild side-effect profile. A phase II trial ARMOR2 is currently recruiting chemo-naïve patients to galeterone (without corticosteroids).
ARN-509 is a second-generation AA that selectively binds to the ligandbinding domain of the AR, blocks its nuclear translocation, and impairs DNA binding to androgen response elements. In preclinical trials, noted to be more potent than enzalutamide, and has less central nervous system penetration. A phase II study included mCRPC chemo-naïve patients, but included patients who had prior abiraterone exposure. At 12 weeks, 88 % and 24 % of the chemo- and abiraterone-naïve patients and 24 % of postabiraterone patients were noted to have PSA responses, respectively. Similarly, median PFS was 19.2 and 8.3 months, respectively.37 Mature data from the above-mentioned phase II trial are not yet available. ARN- 509 is currently being evaluated in combination with abiraterone and also in combination with everolimus in postabiraterone progression.
Several other hormonal agents in development are ODM-201, an AR inhibitor, and VT-464, a novel, nonsteroidal, small-molecule CYP 17A1 inhibitor with selectivity for the lyase activity. Another promising agent in studies is AZD3514 is a first-in-class, orally bio-available drug that inhibits androgen-dependent and -independent AR signaling. It is noted to have activity in advanced CRPC.
The recent approvals of AR-targeted therapies has improved outcomes, broadened the range of options, expanded the therapeutic armamentarium, and raised hope for patients suffering from this terminal disease.
Immune Therapy
Traditionally, PC was felt to be nonimmunogenic, but given the multitude of tumor-associated antigens, such as PSA, prostatic acid phosphatase (PAP), and prostate-specific membrane antigen (PSMA), along with relatively slow growth rate make it a suitable model to explore immunotherapy. The goal is to boost the tumor suppressive response of the patient’s own immune system.
Vaccine-based immunotherapy with Sipuleucel-T (Provenge) was evaluated in minimally symptomatic patients. This is an autologous dendritic cell vaccine, consisting of peripheral blood mononuclear cells, including antigen-presenting cells (APCs) that have been activated ex vivo with a recombinant fusion protein (PA2024) composed of PAP linked to granulocyte-macrophage colony-stimulating factor (GMCSF). In the phase III Identification of Men with a genetic predisposition to ProstAte Cancer: Targeted screening in men at a higher genetic risk and controls (IMPACT) trial, a 4-month improvement in median OS (median survival 25.8 versus 21.7 months) was noted; however, there was no significant effect on the time to objective disease progression or PSA.38 The main side effects are fever, chills, and headache. To explore the question of whether concurrent prednisone use with abiraterone may be sufficiently immunosuppressive to abrogate immunologic response to sipuleucel-T, the P11-3 trial was designed to evaluate the combination of sipuleucel-T and abiraterone acetate plus prednisone. Men with mCRPC were randomized to sipuleucel-T with either concurrent or sequential abiraterone/prednisone. A preliminary analysis demonstrated no difference in the magnitude of immunologic response between the two arms, suggesting that the two therapies may be able to be administered concurrently without loss of efficacy.39 Because the IMPACT trial excluded patients who received docetaxel within the preceding 3 months, the Study for Women With Platinum Resistant Ovarian Cancer Evaluating EC145 in Combination With Doxil® (PROCEED) trial has enrolled men without restriction with regard to prior chemotherapy as part of a phase IV registry.
PROSTVAC-VF, a novel immunotherapy, is a PSA-targeted pox viral-based vaccine, administered with three costimulatory molecules (known as TRICOM)to increase PSA-specific immune responses.
Phase I data showed PSA stabilization in 40 % of patients and, in a phase II study, a 8.5-month improvement in median OS (25.1 months versus 16.6 months) and a 44 % reduction in the death rate (HR 0.56; p=0.0061) was noted.40 In both the phase I and II studies limited toxicity is noted. As the phase I and II data are promising, a randomized placebo-controlled multicenter phase III trial (PROSPECT) is currently ongoing and will evaluate three arms: ProstVac-VF plus adjuvant GM-CSF, ProstVac-VF plus placebo, and placebo-only (ClinicalTrials.gov identifier: NCT01322490).
Using androgen-sensitive and mCRPC cancer cell lines a cell-based vaccine, GVAX, was developed. VITAL-1 and VITAL-2 are phase III trials comparing GVAX with standard docetaxel and in combination with docetaxel, respectively. Neither trials were able to show an OS advantage and were prematurely terminated.
Dendritic-cell-based vaccines in combination with chemotherapy are in clinical trial testing.
Checkpoint modulators of the immune system aim to remove the negative feedback signals in the patient’s own immune system, thereby decreasing the immune system’s tolerance of tumor antigens. These are designed against immune checkpoint molecules present on the T-cell surface such as Cluster of Differentiation 28 (CD28), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and programmed death (PD-1). The blockage of these co-inhibitory signals on the T cell activates the T-cell immune responses. Both ipilimumab and PD-1 inhibitors employ this strategy.
Ipilimumab is a fully humanized monoclonal antibody that blocks cytotoxic T-lymphocyte antigen-4 (CTLA-4). By this, it breaks the immune tolerance by reducing intratumoral regulatory T-cells.41 It is recognized to have an abscopal effect (i.e. immune-mediated anti-tumor activity at a distant site) when combined with local radiation.42
Given this phenomenon, it was evaluated in phase I/II trials, either alone or in combination with radiotherapy (RT) in mCRPC.43 CA 184-043 was a randomized, multicenter phase III trial in mCRPC patients who progressed on docetaxel. It evaluated single-fraction RT to bone metastasis along with ipilimumab. It failed to demonstrate a significant OS benefit (HR 0.85, 95 % CI 0.72–1.00; p=0.053), although there was a notable PFS benefit (HR 0.70, 95 % CI 0.61–0.82; p<0.0001), and improved survival in a prespecified subset with lower disease burden.44
NCT00861614 is another phase III trial in postdocetaxel setting in combination with RT. The study is completed and the results reveal no OS advantage with ipilimumab therapy. Ipilumimab is also studied in the first-line setting, in asymptomatic or minimally symptomatic patients, in a phase III trial NCT01057810, which has completed enrollment. Several studies are underway in combination with ADT in hormone sensitive and resistant settings.
PD-1 is another immune checkpoint receptor expressed by activated T cells, which mediates immunosuppression. An anti-PD-1 antibody (nivolumab) that blocks the binding of PD-1 to PD-L1 (as well as PD-L2) was tested in several solid tumors in a phase I dose-escalation trial with promise, which deserves further exploration.45
Chemotherapy Agents
Until 2010, the standard treatment available for patients who developed castrate resistance or failed secondary hormonal manipulation with a good functional status and end organ function is chemotherapy with docetaxel. The two phase III trials that led to the approval of this agent are TAX 327 and SWOG 9916.
In TAX 327, docetaxel was compared with mitoxantrone, and a 24 % relative risk reduction in mortality and a significant 2.4 months median OS benefit (p=0.009) was noted in the docetaxel arm. Docetaxel was also effective in pain reduction (35 % versus 22 %) (p=0.001).46 In the SWOG study, docetaxel plus estramustine was compared with mitoxantrone plus prednisone, docetaxel conferred a significant survival benefit (HR for death 0.80; 95 % CI 0.67–0.97) and increased median survival (17.5 versus 15.6 months) (p=0.02) over the mitoxantrone arm.47 Hence docetaxel has become the standard of care after progression on hormonal manipulations. But it comes with some limiting toxicities: peripheral neuropathy, cytopenias, and fatigue. Since then, several phase II and III trials were conducted evaluating a combination of docetaxel and several other agents (chemo and non-chemo) and none of them have demonstrated superiority over docetaxel and prednisone.
In the postdocetaxel setting, the use of chemotherapy was unproven until cabazitaxel demonstrated improved survival. The approval of cabazitaxel (see Table 1) was based on the results of the Phase III trial of cabazitaxel for the treatment of metastatic castration-resistant prostate cancer (TROPIC) trial, in which 755 patients whose disease progressed during or after treatment with docetaxel. The cabazitaxel arm showed an improvement in median PFS (2.8 months versus 1.4 months) (p<0.0001), median OS (15.1 months versus 12.7 months), and lower risk for death (HR 0.70) (p<0.0001).48 This 2–3 month improvement in the median OS comes with toxicity from this agent, including significant cytopenias, fatigue, thus requiring dose reductions, and growth factor support. Pooled phase I/II safety data suggested that doses of cabazitaxel <25 mg/m² showed a significantly decreased incidence of neutropenia. A phase III randomized, noninferiority trial, Cabazitaxel at 20 mg/m² Compared to 25 mg/m² With Prednisone for the Treatment of Metastatic Castration Resistant Prostate Cancer (PROSELICA), is currently comparing the efficacy and toxicity of cabazitaxel 20 mg/m² plus prednisone versus cabazitaxel 25 mg/m² plus prednisone in the postdocetaxel setting. Cabazitaxel is also currently studied in the first-line setting, the Cabazitaxel Versus Docetaxel Both With Prednisone in Patients With Metastatic Castration Resistant Prostate Cancer (FIRSTANA) trial is evaluating cabazitaxel versus docetaxel in patients with chemo-naïve mCRPC.49 A randomized phase II study A randomized phase II trial examining an early switch from first-line docetaxel to cabazitaxel, or cabazitaxel to docetaxel, in men with metastatic castration-resistant prostate cancer (mCRPC) (TAXYNERGY), is designed to determine if an earlier switch from one taxane to another is of any clinical benefit. Here patients receive cabazitaxel or docetaxel in the first-line setting. If they do not achieve a ≥30 % PSA reduction after four cycles of treatment, then they will be switched to an alternative taxane agent. It is powered to determine the superiority of an early switch with respect to the primary endpoint of PSA response rate (≥50 % PSA reduction from baseline).50
Radiation Therapy
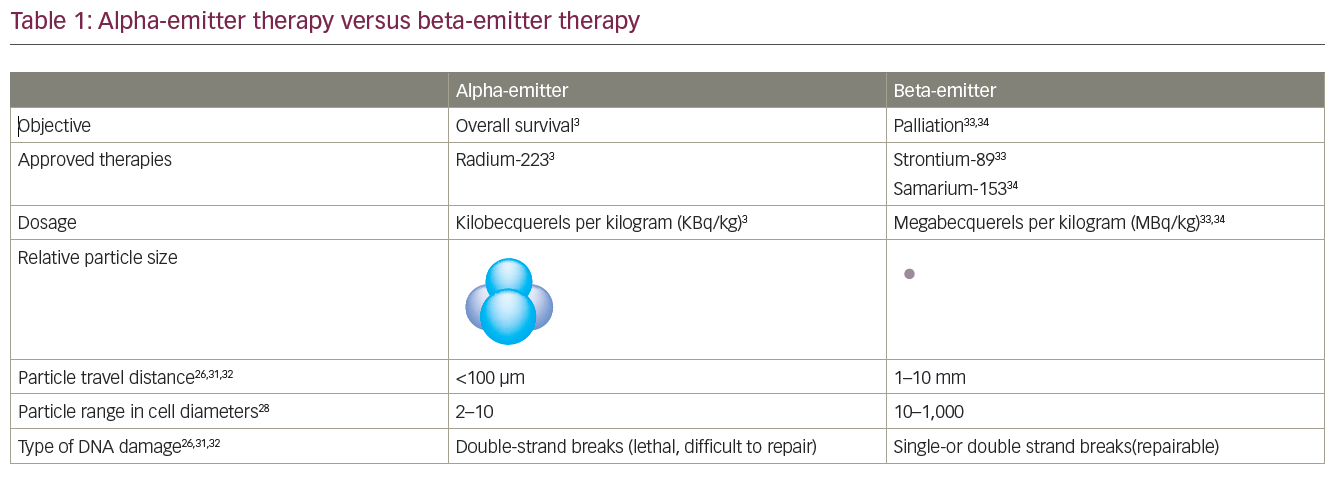
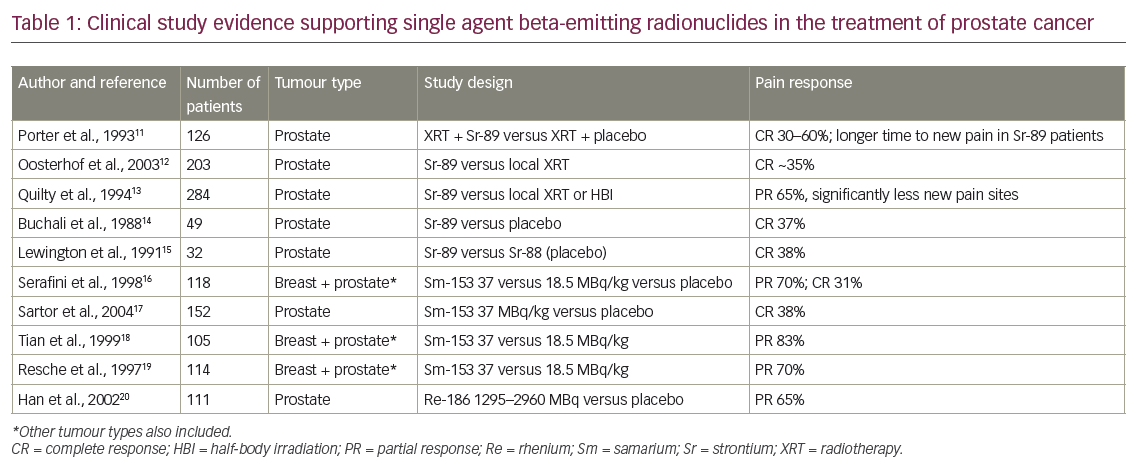
Radiopharmaceuticals have been used for years for pain control in PC. Beta-emitting radioisotopes samarium-153, rhenium-186, and strontium-89 have been shown to reduce pain from bony metastases. But these agents have dose-limiting bone marrow toxicity and limited OS advantage. Unlike the above agents, radium-223 is a novel calcimimetic radiopharmaceutical agent that primarily emits alpha particles. The alpha radiation penetrates relatively shorter distances to deliver higher energy to tumor tissues, leading to double-strand DNA breaks in tumor cells with relatively minimal damage to surrounding normal tissue. Radium-223 is studied in patients who either received, declined, or were not eligible for chemotherapy. In the randomized, placebo-controlled phase III Alpharadin in Symptomatic Prostate Cancer Patients (ALSYMPCA) trial over 900 patients were randomized in a 2:1 fashion. They received six injections of monthly radium-223 (at a dose of 50 kBq/kg of bodyweight intravenous [IV]). About 57 % received docetaxel prior to radium-223 treatment.
It has achieved a survival benefit and also significantly prolonged time to the first symptomatic skeletal event (median 15.6 versus 9.8 months; HR 0.66, 95 % CI 0.52 to 0.83; p<0.001). No significant toxicity or survival difference was noted in patients who received prior docetaxel.51 Based on this, the FDA approved the use of radium-223 in May 2013 for men with symptomatic bone mCRPC without known visceral metastases. Radium-223 is now studied in combination with docetaxel in mCRPC patients with bone metastasis in a phase I/II setting.
Other Novel Agents in Development
A number of novel nonhormonal small molecule inhibitors are currently being investigated in advanced PC patients. Clusterin is a cytoprotective glycoprotein chaperone overexpressed in many cancers. It is induced by stressors such as chemotherapy, radiation, and androgen ablation, AR signaling, etc. It causes treatment-resistance and progression through anti-apoptotic properties.52 Custirsen (OGX-011) is a second-generation antisense oligonucleotide complementary to the clusterin mRNA translation initiation site that forms RNA duplexes and inhibits clusterin expression.This was studied in a phase II trial with docetaxel in men with mCRPC. Patients were randomized to docetaxel plus prednisone with or without custirsen. The addition of custirsen did not appear to add significant toxicity and resulted in self-limited fevers, rigors, and lymphopenia. PSA and objective responses were similar in both arms. A trend towards superior PFS (HR 0.86) and OS (HR 0.61) was noted in the custirsen arm.53 Based on these encouraging results, two phase III trials are currently ongoing. The Comparison of Docetaxel/Prednisone to Docetaxel/Prednisone in Combination With OGX-011 in Men With Prostate Cancer (SYNERGY) trial randomized men with mCRPC to first-line docetaxel plus prednisone with or without custirsen and completed enrollment (NCT01188187). The Comparison of Cabazitaxel/Prednisone Alone or in Combination With Custirsen for 2nd Line Chemotherapy in Prostate Cancer (AFFINITY) trial randomizes men in the second-line setting to cabazitaxel plus prednisone with or without custirsen (NCT01578655).
Tasquinimod is an oral anti-angiogenic quinolone-3-carboxamide derivative with preclinical tumor growth-inhibition properties. Studies have suggested that tasquinimod binds the histone deacetylase HDAC4 to block the action of hypoxia-inducible factor 1-alpha (HIF-1α). Tasquinimod causes induction of thrombospondin-1, an endogenous angiogenesis inhibitor, by downregulation of HIF-1α and vascular endothelial growth factor (VEGF), which in turn leads to reduced angiogenesis via inhibition of the “angiogenic switch,” which could explain tasquinimod’s therapeutic potential.54,55 Tasquinimod was studied in mCRPC patients in a randomized phase II study and it showed a significantly improved PFS in the chemotherapynaïve setting (HR 0.49).56 Twenty-two percent of the patients discontinued treatment secondary to toxicity; 40 % reported grade 3 or 4 toxicities including asymptomatic laboratory abnormalities, such as elevated lipase and anemia; and 4 % were noted to have deep vein thrombosis. A phase III trial in asymptomatic to mildly symptomatic men with mCRPC in the chemonaïve setting has completed enrollment (NCT01234311). Additionally, a phase II proof-of-concept study of tasquinimod maintenance in patients with mCRPC following response or stabilization on first-line docetaxel has completed enrollment (NCT01732549).
Cabozantinib (XL184) is an oral multitargeted receptor tyrosine kinase with predominantly hepatocyte growth factor receptor (MET) and VEGF-inhibition properties. It is currently approved by the FDA for the treatment of progressive metastatic medullary thyroid cancer. In a phase II randomized discontinuation trial, mCRPC patients received cabozantinib for 12 weeks and those without evidence of disease progression were then randomized to either cabozantinib or placebo. The randomization was halted due to early evidence of activity of cabozantinib in terms of improved bone scans and pain. PFS improved significantly compared with the placebo arm (23.9 weeks [95 % CI 10.7–62.4 weeks] versus 5.9 weeks [95 % CI 5.4 to 6.6 weeks] HR 0.12; p<0.001]. The most common grade 3 or greater adverse events included fatigue, hypertension, and hand–foot syndrome.57 Two phase III trials, Study of Cabozantinib (XL184) Versus Prednisone in Men With Metastatic Castration-resistant Prostate Cancer Previously Treated With Docetaxel and Abiraterone (COMET-1) and Study of Cabozantinib (XL184) Versus Mitoxantrone Plus Prednisone in Men With Previously Treated Symptomatic Castration-resistant Prostate Cancer (COMET-2) (see Table 2) have evaluated the role of cabozantinib.
COMET-1 evaluated cabozantinib in men with mCRPC whose disease has progressed on docetaxel as well as abiraterone and/or enzalutamide. Exelixis released the final analysis and the trial did not meet its primary endpoint of a statistically significant improvement in OS. The median OS for the cabozantinib arm was 11.0 months versus 9.8 months for the prednisone arm (HR 0.90, 95 % CI 0.76–1.06; p=0.212). The median PFS was 5.5 months for the cabozantinib arm versus 2.8 months for the prednisone arm (HR 0.50, 95 % CI 0.42–0.60; p<0.0001). The main endpoint of the COMET-2 trial is alleviation of bone pain in mCRPC men with moderate to severe pain despite optimized narcotic medication. This study also enrolled patients with disease progression on docetaxel, abiraterone, and/or enzalutamide. This is also a negative trial and did not meet its primary endpoint—of a pain response at week 6 that was confirmed at week 12 without increase in narcotic medication. Fifteen percent of patients in the cabozantinib arm reported a pain response compared with 17 % in the control arm receiving mitoxantrone/ prednisone. The difference in pain response between the arms was not statistically significant. In the past few years, with extensive research, large strides were made in further understanding of the resistance mechanisms, identifying the critical targetable pathways and development of selective targeted agents was accomplished. But the excitement evoked by these new agents also stimulates a multitude of questions. Is the limited improvement in the survival benefit with all these agents of about 4–6 months explained by acquisition of resistance? The optimal sequence in which to use theseagents has not been established in well-designed randomized trial. The current Enzalutamide With or Without Abiraterone and Prednisone in Treating Patients With Castration-Resistant Metastatic Prostate Cancer (ALLIANCE) trial is evaluating the combination of abiraterone and enzalutamide to enzalutamide alone in mCRPC. The cost of medications and current affordability is a crushing challenge given the expensive nature of these oral medications and the high insurance co-pays. Better practice guidelines are paramount to help patients and oncologists with the judicious selection of therapies, from an expanding armamentarium for an aging population with maintenance of a balance between the risks and potential benefits.