Chronic myelogenous (or myeloid) leukemia (CML) is a myeloproliferative disorder characterized by increased and unregulated growth of granulocytes, leading to high white blood cell counts and splenomegaly. It was estimated that 5,430 adults in the US would be diagnosed with CML in 2012 and 610 would die of the disease.1 In the absence of intervention, CML typically begins in the chronic phase, and over the course of several years progresses to an accelerated phase and finally to a blast or acute phase. The latter is the terminal phase of CML and clinically behaves like an acute leukemia. Until recently, interferon alpha (IFN-α) and allogeneic stem cell transplantation (SCT) formed the mainstay of treatment in CML. However, the utility of both is limited by adverse effects (AEs).2,3
CML is associated with a characteristic chromosomal translocation called the Philadelphia chromosome, which results in the expression of a tyrosine kinase molecule: the breakpoint cluster region–Abelson (BCR-ABL) protein.4 The high prevalence of this protein makes it an attractive molecular target for therapeutic approaches to CML. In the last decade, the development of tyrosine kinase inhibitors (TKIs) that inhibit signaling on the BCR–ABL protein has dramatically improved outcomes for patients with CML. Before the US Food and Drug Administration (FDA) approval of imatinib in 2003, median survival was around 4 to 5 years from diagnosis. Current estimates of life expectancy in patients that respond to TKIs are similar to that of the general population.5
However, despite the success of TKIs, drug resistance is an unmet need in the treatment of CML. Furthermore, full molecular remission (MoR) (i.e. to become negative for BCR-ABL) is rarely achieved with TKI inhibitors. This article aims to review the use of TKIs and examine alternative therapeutic targets with the potential to eradicate minimal residual disease (MRD).
Available Tyrosine Kinase Inhibitor Therapies for Chronic Myeloid Leukemia
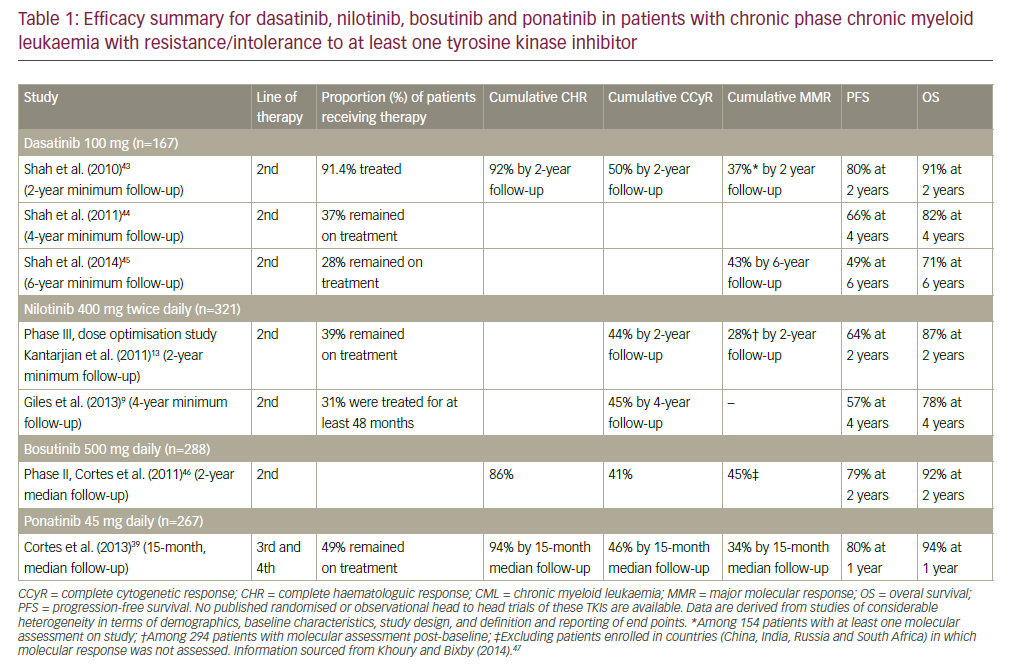
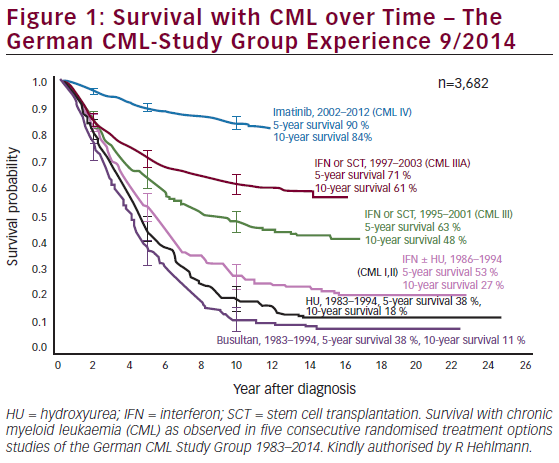
Current FDA-approved treatment options for CML are summarized in Table 1. Based on the results of the International Randomized Study of Interferon and STI571 (IRIS) trial in 2003, in which imatinib demonstrated superiority over IFN-α plus low-dose cytarabine in terms of hematologic and cytogenetic responses, tolerability, and the likelihood of progression to accelerated-phase or blast-crisis CML,6 imatinib replaced IFN-α as the standard of care in CML. It has achieved rates of complete cytogenetic response (CCyR) of more than 40 % in patients after failure of IFN and more than 80 % in newly diagnosed patients.7 Its use has been associated with positive long-term outcomes.8,9 However, significant proportions (10– 20 %) of patients do not achieve a satisfactory response to imatinib and discontinue therapy; a further 10–15 % will achieve a satisfactory response but subsequently acquire resistance to imatinib.7,10,11 A significant number of patients develop AEs on imatinib therapy that cannot be managed through dose reduction or symptomatic treatment.11 Common AEs include neutropenia, thrombocytopenia, anemia, and elevated liver enzymes.12 Uncommon or delayed AEs may include renal and dermatologic problems.13 imatinib has also been associated with cardiac adverse reactions related to c-ABL inhibition, suggesting that such AEs may be common to all TKIs.14 Response rates and the durability of responses to imatinib are dependent on the stage of disease at which treatment is initiated (see Figure 1).7
The second-generation TKIs dasatinib and nilotinib have higher binding affinity than imatinib to kinase domains of BCR-ABL and received FDA approval as second-line treatments in 2006 and 2007, respectively, and in the first-line treatment setting in 2010, after clinical trials data demonstrated their superior efficacy and safety.15,16 However, in spite of the advantages of dasatinib and nilotinib, treatment with these agents has been associated with AEs requiring dose interruptions and reductions.17,18 In 2012, bosutinib, a second-generation inhibitor of ABL and Src family kinases, was approved by the FDA as a second-line treatment option for adult patients with previously treated CML, following two phase I/II clinical trials.19,20 Bosutinib inhibits wild-type BCR-ABL and most imatinibresistant BCR-ABL mutations, except for V299L, F317V, and T315I, and is characterized by low hematologic toxicity and few AEs. Bosutinib has also demonstrated efficacy in the third-line treatment setting.19 However, in a phase III trial that compared bosutinib with imatinib in the first-line treatment setting, bosutinib did not meet its primary end-point of CCR at 12 months, despite the observed higher major molecular response (MMR) rate at 12 months, faster times to CCR and MMR, fewer on-treatment transformations to accelerated/blast phase, and fewer CML-related deaths with bosutinib compared with imatinib.21
Despite these advances, there remains a small subset of patients who do not respond to TKIs. The mechanisms of drug resistance are diverse, but, in most cases, mutations are found at the time of resistance that change amino acids within the kinase domain of BCR-ABL T315I gene. This confers resistance to the first- and second-generation TKIs.10 The T315I mutation is present in up to 40 % of TKI-resistant patients22–24 and, until recently, has been resistant to all approved therapies.25 The only treatment option for patients with the BCR-ABL-T315I mutation used to be SCT.26–28
The third-generation TKI ponatinib is a BCR-ABL inhibitor that was approved by the FDA in December 2012 for the treatment for the treatment of adult patients with chronic phase, accelerated phase, or blast phase CML that is resistant or intolerant to prior TKI therapy. It has demonstrated significant efficacy in CML patients, including those with the T315I mutation.4 In the 12-month follow-up of the phase II Ponatinib Ph ALL and CML Evaluation (PACE) trial, responses occurred early and were durable: of patients with chronic phase CML, 98 % had a complete hematologic response, 72 % had a major cytogenetic response (MCyR), and 44 % had a MMR.29,30 However, the FDA issued a black box warning about potential AEs including arterial thrombosis and liver toxicity.31 Due to the high prevalence of AEs, the FDA suspended the use of the drug as of November 2013.
In addition to the problem of drug resistance, nonadherence to BCR-ABL inhibitors has been observed in more than one-third of CML patients, and 100 % adherence is rare. Furthermore, reduced adherence to BCR-ABL inhibitors is associated with reduced efficacy.32,33
Another concern in the use of TKI inhibitors is MRD. The total number of leukemia cells in the body is reduced substantially in patients with BCRABL- positive CML responding to imatinib, but they are not eradicated— only a small proportion of patients in the IRIS trial achieved MoR.34 MRD usually remains detectable through molecular monitoring of BCR-ABL transcript levels with reverse transcription-polymerase chain reaction (RTPCR), indicating that disease eradication with TKI inhibitors may pose a significant challenge.7 MRD is thought to reside in TKI-insensitive leukemia stem cells (LSCs),22 and largely results from mutations in the kinase domain of the BCR-ABL gene that impair imatinib binding. Such mutations have been found in 60 % of patients who experience imatinib resistance.35 Studies of imatinib-treated patients suggest that the BCR-ABL levels measured early in therapy may predict durable cytogenetic remission and thus progression free-survival (PFS) or acquisition of resistance.7
The definition of outcome-associated landmarks during TKI treatment is required to improve the survival of CML patients.36 The importance of an early MMR for overall survival (OS) has not been fully established, although significantly improved survival rates for patients with BCR transcript levels >9.84 % and 10 % at 3 months have been reported.37,38 Regular monitoring of MRD enables the identification of those with suboptimal response and enables early therapy switching.39 For these patients, a switch in TKI for is recommended as early as 6 months after first-line TKI initiation.40,41 In a study of 123 patients with CML who were treated with second-generation TKIs after imatinib failure, it was found that achievement of a 3-month CCR was the only predictor of event-free survival and OS in patients receiving second line of therapy after imatinib failure. Patients who fail to achieve CCR may not obtain long-term benefit from TKI therapy.42 In order to achieve the goal of curing CML, it is necessary to eradicate MRD. However, currently available TKI inhibitors are not capable of eliminating MRD. Alternative treatment options capable of targeting LSCs are therefore required.
Alternatives to Tyrosine Kinase Inhibitors
Omacetaxine
The potential for the elimination of CML cells, including LSCs, through simultaneous inhibition of BCR-ABL and other molecular targets, has increased through following advances in CML biology and drug therapy.43 Omacetaxine mepesuccinate (Synribo®, Teva) is a derivative of homoharringtonine, a natural plant alkaloid, and provides a unique approach to the treatment of relapsed and refractory CML whose mechanism of action is independent of tyrosine kinase. It does not require binding to BCR-ABL and is not affected by resistance-conferring mutations in the BCR-ABL gene.44,45 Omacetaxine, a reversible protein translation inhibitor, reduces levels of multiple oncoproteins, including BCR-ABL, and induces apoptosis in LSCs.45 It has clinical activity against resistant CML, including cells containing the T315I mutation.46,47 Its precise mechanism of action is not known, though several potential pathways are thought to be involved. Omacetaxine reduces the expression of BCR-ABL both in vitro and in animal models,44 possibly a result of its effect on heat-shock protein 90 (Hsp90).48 It also downregulates myeloid leukemia cell differentiation protein (MCL-1), which is an important short-lived anti-apoptotic BCL-2 family protein. In addition, its clinical efficacy may be partly due to its inhibitory activity on LSCs.44 It was developed in the 1980s but its clinical development was interrupted by the introduction of imatinib.
Clinical data indicate that omacetaxine has the potential to reduce MRD: in an in vitro model it killed 90 % of LSCs.44 Furthermore, omacetaxine prolonged survival in animals carrying the T315I mutation (see Figure 2).44 Homoharringtonine and imatinib have synergistic or additive effects in CML cell lines.49 Preliminary data in patients with CML with T315I mutations who were resistant to imatinib showed a complete hematologic response in 85 % and a 57 % reduction in the T315I clone.50 These data suggest that the combination of imatinib and omacetaxine warrants further investigation.
Two international phase II clinical studies (CML-202 and CML-203) have demonstrated the efficacy of omacetaxine as second- and laterline therapy in CML patients. The first (n=22) assessed the efficacy of omacetaxine after TKI failure in patients with chronic-phase CML with T315I mutation.51 Complete hematologic response was reported in 77 % of patients (95 % lower confidence limit, 65 %); median response duration was 9.1 months (see Figure 2). MCyR was seen in 23 % (95 % lower confidence limit, 13%), including CCR in 16 %. Median PFS was 7.7 months. Omacetaxine was found to have a manageable safety profile that primarily comprised predictable and reversible myelosuppression, a common effect of antileukemic therapies. Grade 3/4 hematologic toxicity included thrombocytopenia (76 %), neutropenia (44 %), and anemia (39 %), and was successfully managed by dose reduction. Nonhematologic AEs were mostly mild to moderate in severity and included infection (42 %), diarrhea (40 %), and nausea (34 %).
The second trial assessed omacetaxine in patients with chronic-phase CML and resistance or intolerance to two or more TKIs.52 Patients (n=46) received subcutaneous omacetaxine 1.25 mg/m2 twice daily days 1–14 every 28 days until hematologic response (up to a maximum of six cycles), then days 1–7 every 28 days as maintenance. Hematologic response was achieved or maintained in 67 % of patients, the median response duration was 7.0 months. A MCyR was reported in 22 % including CCR in 4 %. Median PFS was 7.0 months (95 % confidence interval [CI], 5.9–8.9 months), and OS was 30.1 months (95 % CI, 20.3 months–not reached). Grade 3/4 hematologic toxicity included thrombocytopenia (54 %), neutropenia (48 %), and anemia (33 %).
Omacetaxine received FDA approval in October 2012 for the treatment of adult patients with chronic or accelerated phase CML with resistance and/or intolerance to two or more TKIs. Approval was based on an FDA review of data from 111 patients with CML who had received two or more prior TKIs, including imatinib. Major cytogenetic response was achieved in 18 % of patients with chronic phase CML, with a median response duration of 12.5 months. Major hematologic response was achieved in 14 % of patients with AP, with a median response duration of 4.7 months.53 Since mutation testing is widely available for patients with CML, omacetaxine represents an important treatment option in patients with chronic-phase CML with T315I mutation, who otherwise have a poor prognosis.
lnterferon-α
Before the advent of imatinib, IFN was the mainstay of CML therapy, and there has been a recent resurgence of interest in this therapy. IFN-α has a broad range of therapeutic effects that may reduce resistance or relapse, especially when used in combination with other CML therapies. IFN has important immunomodulatory properties, and a possible role in eradicating the LSC, making it potentially useful for the eradication of MRD. Pegylated IFN (Peg-IFN-α) is better tolerated than regular IFN-α and its use for the elimination of MRD has been studied, both as add-on therapy to TKI agents for those who have not achieved a complete molecular response to TKI therapy alone,54 At the 2012 American Society of Hematology (ASH) conference, data were presented from the French SPIRIT study, which showed that the combination of imatinib with Peg-IFN-α can trigger a faster and deeper molecular response than an imatinib monotherapy.55 The SPIRIT research group also presented preliminary data from a single-arm study that investigated the combination of nilotinib with Peg IFN-α.56 Newly diagnosed CML patients first received Peg IFN-α alone for 1 month, followed by a halfdose of IFN-α combined with 2 x 300 mg of nilotinib. At month 12, over 50 % had achieved a molecular response and another 24 % reached MMR, meaning that three out of every four patients were in good remission after 12 months. Commonly reported AEs included anemia in 5 % of patients and thrombocytopenia among 41 %, but these tapered off. Furthermore, during the first 3 months, alterations in liver metabolism and serum lipid levels were reported, as well as stomach pains and depression. A French phase III study is being planned. The Tasigna and Interferon Alpha Evaluation Initiated by the German Chronic Myeloid Leukemia Study Group (TIGER) study, which will compare nilotinib with nilotinib + Peg IFN-α combination, is currently enrolling. Other studies aim to predict early response to IFN-α and the use of IFN-α in patients with the T315I mutation.57
Alternative Molecular Targets in Chronic Myeloid Leukemia
In addition to omacetaxine and interferon, several other therapies that target alternative molecular targets are undergoing clinical investigation (see Figure 3). Drugs currently in clinical development are summarized in Table 2. Aberrant DNA methylation is associated with disease progression, resistance to imatinib, and shortened survival in CML, providing a rationale for the use of hypomethylating agents.58,59 Decitabine, a cytosine that incorporates into DNA and depletes DNA methyltransferase levels, has demonstrated clinical activity in CML, including imatinib-resistant cases.60 In addition, combination therapy with decitabine and imatinib has been shown to be well tolerated and active in advanced phase CML without BCR-ABL kinase mutations.60 The efficacy of the combined treatment depends on residual sensitivity to imatinib.61,62
Activation of the gene regulatory factor STAT5 is critical for the maintenance of CML characterized by BCR-ABL. This has led to clinical research into TKIs for the STAT5-activating janus kinase 2 (JAK2). The first JAK2 TKI, ruxolitinib, has been approved by the FDA for use in patients with myelofibrosis, and several JAK2 TKIs are in clinical development for CML.63–65 Recent data suggest that JAK2 alone is not a drug target in CML.66 However, a recent study suggests that simultaneously targeting BCR-ABL and JAK2 activities in primary CML stem/progenitor cells may improve outcomes in patients that develop resistance to imatinib.67
An accumulating body of clinical data also suggests a role for hedgehog (HH) signaling, including the smoothened transmembrane protein (Smo), in the maintenance and proliferation of LSCs in CML.68,69 The combination of nilotinib plus the Smo inhibitor LDE225 has demonstrated inhibition of primitive CML stem cells.70 Thus the combination of HH and TKI inhibitors warrants further investigation.
in vitro and in vivo studies suggest that the arachidonate 5-lioxygenase (5-LO) gene (Alox5) is involved in the development of CML.71–73 Zileuton is an inhibitor of 5-LO, the product of the Alox5 gene, and has shown promising efficacy in animal studies.73
Hsp90, a chaperone of several oncoproteins including BCR-ABL, is another potential therapeutic target in CML.74 Hsp90 inhibitors can inhibit the ability of the protein to function as a chaperone, thereby downregulating of BCR-ABL mutants including T315I mutants and can also induce apoptosis in CML cell lines.75 An Hsp inhibitor, IPI-504, prolonged survival of mice with BCR-ABL-T315I–induced leukemia and suppressed LSCs.76 Analogs of geldanamycin, a naturally occurring Hsp90 inhibitor, are also in clinical development.77
The programmed cell death 1 (PD-1) pathway is emerging as an important tumor evasion pathway in numerous cancers.78 Blocking PD-1 prolonged survival in animal models.79 In addition, PD-1 is upregulated on CD8+ T cells from CML patients.79 These data suggest that blocking the PD-1 may represent a novel therapeutic approach for CML.
Other therapeutic options under clinical investigation for CML include dual Src-family kinase/ABL kinase inhibitors. Src kinases are important mediators of downstream signaling by ABL from cell-surface receptors. Therefore, inhibitors of these enzymes may act synergistically with BCR-ABL inhibitors and hence potentially inhibit alternative survival pathways. Bosutinib is a dual inhibitor of both Src and ABL kinases.80 Bafetinib (INNO-406), a dual Abl/ Lyn kinase inhibitor, has also demonstrated efficacy in early clinical studies.81 The Aurora family of serine/threonine kinases are essential for cell proliferation and are expressed in numerous cancers.25 Aurora kinase inhibitors in clinical development for CML include tozasertib82 and danusertib.83
Histone deacytylases control the coiling and uncoiling of DNA necessary for gene expression. Vorinostat, a histone deacytylase inhibitor, has shown efficacy in acute leukemia in combination with imatinib84 and nilotinib.85A recent study found that signals from the bone marrow microenvironment protect CML LSCs from TKI treatment through N-cadherin and Wntb- catenin signaling, providing a new potential target for therapeutic intervention.86 Other potential therapeutic agents include proteasome inhibitors and cyclin-dependent kinase inhibitors.48,87
Several vaccines are also being studied for use in CML. Results from a small multicenter observational trial (n=10) indicated that addition of the multipeptide vaccine CMLVAX100 to imatinib treatment in patients with CML may reduce MRD and increase the proportion of patients achieving a MMR.88 The PR1 vaccine, which is derived from two myeloid leukemiaassociated antigens, has demonstrated efficacy in clinical studies.89 However, subsequent clinical trials involving peptide vaccines have yielded limited success,90,91 and current research is focused on DNA vaccination.92,93
Summary and Concluding Remarks
It is clear that, despite the success of TKI therapy, unmet clinical needs remain in the treatment of CML. Furthermore, clinical evidence suggests that TKI therapy is insufficient to eradicate MRD. Therefore, alternative therapeutic approaches are required, which may involve inhibitors of non-BCR-ABL targets or targets downstream of BCL-ABL to prevent or overcome resistance.
Research has also focused on therapies that target alternative pathways. Future studies should investigate the addition of omacetaxine to TKIs in patients with CML with the aim of achieving CCR in persistent molecular disease. Concerns persist regarding the administration route of omacetaxine, which is not currently approved for self-administration despite the fact that all clinical studies with subcutaneous omacetaxine were self-administered without any safety concerns. Allowing for patient self-administration will improve access and compliance, broaden the use of omacetaxine, and reduce the cost of care. The resurgence in interest in IFN-α2β has also led to promising clinical trial data. IFN-α2β has a broad range of therapeutic effects that may reduce the likelihood of resistance or relapse, especially when used in combination with other CML therapies.
The increasing knowledge about the biology of CML and clinical research into new therapeutic targets will broaden the treatment strategies used for the individual patient with CML in the future, and eradication of MRD has become a realistic target.