Acute myeloid leukaemia (AML) is a heterogeneous clonal disorder of haematopoietic progenitor cells resulting in uncontrolled growth and accumulation of malignant white blood cells. It is the most common myeloid leukaemia in adults, with a prevalence of 3–8 cases per 100,000 adults rising to 9–17 cases per 100,000 adults aged 65 years and older. The median age at presentation is about 70 years.1 AML affects both male as well as female patients with a slight predominance of the male gender (m/f: 3:2). According to the American Cancer Society, AML accounted for approximately 33% of all new leukaemia cases in the United States in 2016. Almost 20,000 patients had been newly diagnosed with AML in 2016 in the United States and over 10,000 died of the disease (www.cancer.org). The median overall survival (OS) after 5 years in younger (18–60 years) adult AML patients is roughly 40% with the disease being even more detrimental in older individuals with only around 10% surviving patients above the age of 60 years.2 Hence, there is a high medical need to improve the outcome of AML patients.
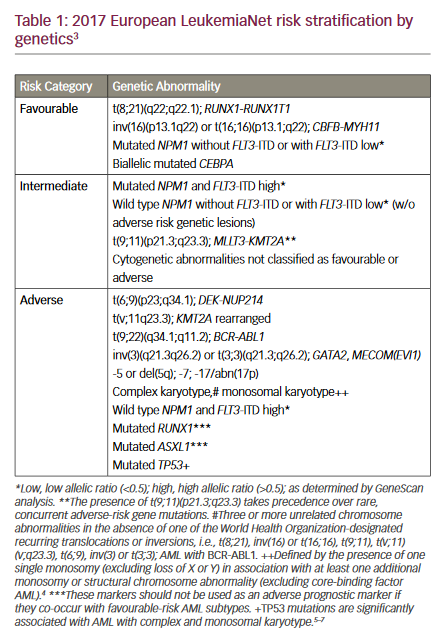
The prognosis for patients with AML is determined to a large degree by the biology of the disease. In recent years, the identification and characterisation of genetic aberrations has vastly improved our understanding of the pathogenesis of AML. These genetic alterations allow for the stratification of patient populations into different risk groups, thus guiding treatment. Based on the currently updated version of the European LeukemiaNet (ELN) risk stratification by genetics, the risk groups consist of the favourable, intermediate and adverse risk categories (Table 1).3–7
AML with normal karyotype (accounting for roughly 50% of the patients) can be categorised according to molecular abnormalities. Of these, the most frequently affected gene mutations are NPM1 and FLT3.8
Despite the fact that AML is a clinically and genetically heterogeneous disease, until recently most patients have been treated by similar chemotherapeutic regimens.9 To date, the only approved targeted therapies for patients with AML are all-trans retinoic acid10 and arsenic trioxide11 for acute promyelocytic leukaemia (APL), which accounts for 10–15% of AML cases.12
There is a clear need for more targeted therapies and a more individualised approach in the treatment of AML. However, in the last decade the treatment options for AML have expanded as a result of the discovery of cytogenetic abnormalities as well as molecular mutations, but only two new nontargeted drugs have been approved in the EU in this period. This article aims to discuss mutations of the FLT3 gene, as well as the therapeutic interventions targeting these mutations.
Activating mutations in acute myeloid leukaemia
A number of cytogenetic abnormalities, mutations or epigenetic alterations, which are involved in the pathogenesis of leukaemia, have been identified (Figure 1).13
Activating mutations of FLT3, resulting in the constitutive activation of this receptor tyrosine kinase, are among the most frequent molecular abnormalities in AML and are present in about 30% of newly diagnosed patients.14,15 FLT3 is a member of the class III receptor tyrosine kinase family and has an established role in normal growth and differentiation of haematopoietic precursor cells.16 Following ligand binding, the FLT3 receptor dimerizes at the plasma membrane, leading to a conformational change in its activation loop that allows adenosine triphosphate (ATP) access to the FLT3 active site. This is followed by autophosphorylation and activation of numerous downstream signalling pathways.17–19 Mutations of the FLT3 gene lead to ligand-independent activation and dysregulation of downstream pathways such as PI3K/AKT, MAPK/ERK and STAT5.20–22 These pathways inhibit apoptosis and differentiation, and promote proliferation. The frequency of mutated FLT3 in AML, its location on the cell surface and its association with an adverse prognosis make it an attractive target.23
Internal tandem duplications
The most common FLT3 mutations are internal tandem duplications (ITDs), which occur in roughly 20–30% of all AML patients, particularly, not only in cytogenetically normal AML,13,24 but also in APL with t(15;17) (q22;q12) and AML with t(6;9)(p23;q34). Its incidence is associated with age: it can only be rarely found in children, whereas its incidence is highest in young adults up to the age of 60 years and declines in

the elderly.24 Clinically, FLT3-ITD mutations are associated with a high white blood cell count, a high percentage of myeloid blast cells in the peripheral blood and bone marrow, and a more frequent diagnosis of de novo rather than secondary AML.25,20
In cytogenetically normal AML the presence of an ITD is associated with an increased relapse rate and reduced OS as compared to wild type FLT even after allogeneic haematopoietic stem cell transplantation (allo HSCT).28 FLT3-ITD is also a negative prognostic factor for survival in patients with either refractory or relapsed AML as they have a poor response to salvage therapy.29–32
Regarding specific ITD characteristics, the size of these duplications varies widely, typically ranging from 3 to over 100 base pairs (bps) with a median of 48 bps.33 In addition, size and ITD insertion site in the FLT3 gene seem to be correlated in that the more 3s the insertion site, the longer the ITD is.33 The impact of the size on outcome is still unclear with some publications stating that there is no impact on outcome,34,35 whereas one publication found that short ITDs may impart an unfavourable outcome.36 Nevertheless, most publications stated that longer ITDs correlate with lower complete response (CR) rates and shorter OS and event-free survival (EFS).37–39
Recently, the ITD insertion site within the FLT3 gene has been shown to be an important prognostic factor.33 About three-quarters of FLT3-ITDs occur in the juxtamembrane domain (JMD) whereas one-quarter in the tyrosine kinase domain 1 (TKD1) of the FLT3 gene, particularly in the ß1-sheet.33 In cell culture analyses, a prototypic FLT3-ITD with insertion site in the ß2-sheet of the TKD1 (FLT3-ITD627E) mediated phosphorylation of FLT3 and STAT5, suggesting that non-JMD FLT3-ITD mutations confer constitutive activation of the receptor.40 In addition, FLT3-ITD627E induced transformation of haematopoietic 32D cells and led to a lethal myeloproliferative disease in a syngeneic mouse model. Insertions in the ß1-sheet of TKD1 may introduce a greater instability into the protein structure and may therefore be associated with a pronounced adverse prognosis.33,40 In DNA-based Sanger sequencing analysis of diagnostic samples from 241 FLT3-ITD mutated patients, an ITD insertion site in the ß1-sheet of the TKD1 was associated with an inferior prognosis as compared to other insertions sites in terms of achievement of complete remission (CR; odds ratio [OR], 0.22; p=0.01), relapse-free survival (RFS; hazard ratio [HR], 1.86; p<0.001) and OS (HR, 1.59; p=0.008).33
Besides the insertion site, further prognostic and predictive impact has been shown for the allelic ratio, which is quantified by GeneScan analysis using DNA fragment analysis.41,42 This method is a semi-quantitative assessment of the FLT3-ITD allelic ratio, expressing the allelic ratio as a percentage of the area under the curve for FLT3-ITD divided by the area under the curve for wild-type FLT3. According to different publications the distribution of the allelic ratio varies widely.33,34,39,42,43 Therefore, the question arises where the optimal cut-off value should be to distinguish patients with high versus low allelic ratio. Currently, there is still a lack of consensus on clinically relevant cut-offs between high/low allelic ratios, which might be due to different methodologies of testing that had been applied. Nevertheless, there is a clear association of an inferior OS and EFS in patients with higher allelic ratios.33,38,42,44 In addition, as AML evolves from diagnosis to relapse, the allelic ratio seems to increase.45,46 However, in a small proportion of relapses FLT3-ITD was no longer detectable.47
Point mutations of the tyrosine kinase domain In addition, approximately 5–10% of AML patients harbour point mutations within exon 20 of the FLT3 gene (FLT3-TKD).22 TKD mutations most frequently occur at codon 835 where a tyrosine residue replaces aspartic acid, stabilising the activation loop in the ATP-bound configuration and promoting activation.48 Other point mutations include, for instance, N676D, I836S and Y842C in the TKD1 and TKD2 domains, respectively.49 Unlike ITDs, the incidence of point mutations is not associated with age50 and their prognostic significance is discussed controversially.14,44,48,51–54 Nevertheless, TKD mutations can occur after treatment with tyrosine kinase inhibitor (TKI) as a mechanism of resistance, thus implicating an adverse prognosis.55–57
Concurrent mutations
The prognostic impact of FLT3-ITD is also affected by concurrent mutations, such as nucleophosmin 1 (NPM1) and DNMT3A. In normal karyotype AML (CN-AML) with NPM1 mutation, FLT3-ITDs are present in about 45% of patients.58,59 Mutations in exon 12 of the NPM1 gene cause cytoplasmic dislocation of the NPM1 protein.60 As a result, cytoplasmic NPM1 is unable to undertake its normal functions as binding and transporter protein. In CN-AML, NPM1 mutations without FLT3-ITD59 or a low allelic ratio are a more favourable prognostic factor.42
The prognostic effect of concurrent FLT3 and NPM1 mutations is a matter of controversy. A cohort study of young adult AML patients identified three prognostic groups: good (FLT3-ITD[-]NPM1[+]); intermediate (FLT3– ITD[-]NPM1[-] or FLT3-ITD[+]NPM1[+]) and poor (FLT3-ITD[+]NPM1[-]). The authors concluded that patients with an FLT3-ITD mutation burden greater than 50% or (FLT3-ITD[+]NPM1[-]) have a poor prognosis and may be good candidates for experimental therapeutic approaches.61 However, another study found that the FLT3-ITD load also has to be taken into account: in patients with a high FLT3-ITD allelic burden, the effect of an NPM1 was less important.62 A study of older AML patients suggested that NPM1(+)/FLT3-ITD–(-) confers a favourable prognosis for patients with AML of ages 55–65 years but not in those of age >65 years.63 Recent recommendations from the ELN include a revised version of the risk stratification according to genetics including the allelic ratio (Table 1).3
Detection of minimal residual disease
Pretherapeutic molecular testing for NPM1 and FLT3 is considered a standard of care to determine the best treatment option. Whereas NPM1 has been shown to be a reliable marker for minimal residual disease (MRD) detection with high sensitivity,64–66 the suitability of FLT3-ITD for MRD detection has been questioned. First, FLT3-ITD mutations display substantial heterogeneity in terms of size, number of clones per patient, allelic ratio and insertion site within the FLT3 gene and second, its proposed instability (reported on about 25% of paired diagnosis-relapse samples) during the course of treatment. Current methods used to determine FLT3-ITD mutations have limited sensitivity and are not suitable for MRD detection. Newer techniques, such as real-time quantitative polymerase chain reaction (RT-qPCR) with patient-specific primers, aim to improve the sensitivity of FLT3-ITD.67 However, this approach has limitations, since each FLT3-ITD mutation needs a clone-specific primer/probe set, which is time-consuming and may not be possible in every case. In addition, direct sequencing may not be possible in patients with a low allelic ratio since the wild-type sequence is competitively amplified. Recently, another PCR-based assay for FLT3-ITD MRD was reported.68,69 This assay employed primers oriented in the opposite direction; hence, amplification occurred only if an FLT3-ITD was present. Again, this approach has limitations, since short FLT3-ITDs (less than 30–40 bases) are not detected due to insufficient primer annealing space, which may apply to roughly 25% of all FLT3-ITD cases. Both approaches are therefore not ready to be implemented in clinical routine care. Next-generation sequencing (NGS) is potentially useful70 but generates complex data which is still expensive and requires considerable expertise to interpret.
Nevertheless, in those patients with a concurrent NPM1 mutation, MRD can be assessed by analysis of NPM1 mutated transcripts.66
In summary, FLT3 mutational testing should be mandatory in all AML patients at diagnosis as well as at relapse for prognostic purposes and for guiding therapeutic decisions. At present, it has little utility for MRD monitoring until different methodologies can be standardised.
Treatment options for FLT3-ITD acute myeloid leukaemia
In younger patients with newly diagnosed AML considered suitable for intensive induction therapy, the combination of an anthracycline and cytarabine (“7+3” regimen) remains the standard of care also for patients with activating FLT3 mutations.71 However, higher allelic ratios were associated with lower CR-rates after induction therapy42 bringing up the question of dose-intensification in these patients.72,73 In older adults a substantial proportion of patients cannot tolerate intensive induction chemotherapy; in these cases other less intensive regimens may be used including low-dose cytarabine74 or hypomethylating agents (e.g., azacitidine or decitabine).75,76 Based on the assessment of the risk–benefit ratio (i.e., non-relapse mortality/morbidity versus reduction of relapse risk) allo HSCT from matched-related or unrelated donor in early first CR is the treatment option for patients with intermediate and adverse risk genetics. In addition, allo HSCT has been shown to improve outcomes in FLT3-ITD AML, particularly in patients with a high allelic ratio.77–79 Nevertheless, recent studies indicate that AML patients with NPM1 mutation and low FLT3-ITD allelic ratio may have a more favourable prognosis and should therefore not routinely be assigned to allo HSCT.42,43,80 In contrast, an ITD insertion site in the TKD1 remained an unfavourable prognostic factor regardless of the applied therapy.42 Another important prognostic factor has been shown for the NPM1 MRD status after the second chemotherapy66 or before allo HSCT.66,81
New therapies targeting FLT3
In the last decade, numerous small molecule FLT3-TKIs have been developed to disrupt oncogenic signalling. Most compete for the ATP binding site in the active domain of the kinase, inhibiting protein phosphorylation.82
Early TKIs, rather than being specifically designed to target FLT3, had multiple targets including KIT, PDGFR, VEGFR and JAK2.83 Several agents have shown evidence of modest antileukemic activity as monotherapy including midostaurin (phase IIb),84 linifanib (phase I),85 semaxanib (phase II)86,87 tandutinib88 and KW-2449 (preclinical).89 However, responses were typically short-lived and mostly partial remissions.
Moreover, it has been suggested that the responsiveness to FLT3 TKIs seems to depend on the FLT3 allelic ratio.46 In an in vitro analysis, six different FLT3 inhibitors (lestaurtinib, midostaurin, AC220, KW-2449, sorafenib and sunitinib) were examined for potency against mutant and wild-type FLT3, as well as for cytotoxic effect against a series of primary blast samples obtained from FLT3-ITD mutated AML patients. Relapsed samples and samples with a high allelic ratio were more likely to be responsive to cytotoxicity from FLT3 inhibition as compared to the samples obtained at diagnosis or those with a low allelic ratio.46 Therefore, it has been hypothesised that patients with newly diagnosed FLT3-mutant AML might be less likely to respond to highly selective FLT3 inhibition.46 However, the results probably indicate that the presence of an FLT3-ITD with even a low allelic ratio cannot be excluded altogether from prognostic risk stratification.
Results of clinical trials with tyrosine kinase inhibitor treatment
The major clinical studies investigating TKI treatment in FLT3-mutated AML are summarised in Table 2. In a phase I/II study, monotherapy with lestaurtinib demonstrated biologic and clinical activity in five out of 14 heavily pretreated patients with relapsed or refractory FLT3– mutated AML, including reductions of blast cells from bone marrow and peripheral blood.90 In addition, in a phase II trial, single agent lestaurtinib has shown modest activity as first-line treatment for older AML patients who were unfit for intensive chemotherapy. Within this trial, lestaurtinib was given orally for 8 weeks, starting with 60 mg twice daily (bid), escalating to 80 mg bid, and was generally well tolerated. Clinical activity included transient reductions in bone marrow and peripheral blast cells in three of five patients with mutated FLT3 and five of 22 evaluable wild-type FLT3 patients.91 In both studies, FLT3 inhibition correlated with clinical response. These findings prompted a large, multicentre phase III clinical trial evaluating lestaurtinib in combination with chemotherapy in relapsed/refractory patients. However, no increase in response rates or prolongation of survival of AML patients with activating FLT3 mutations was found.92,93 In addition, it has been shown that plasma FLT3 ligand levels rise dramatically after chemotherapy and this has been suggested to interfere with the bioavailability of FLT3 TKIs.94 This issue has been evaluated in a meta-analysis of two consecutive phase III trials of the Medical Research Council (AML15 and AML17 trials), including n=500 FLT3-mutated AML patients. Within this trial, newly diagnosed AML patients with activating FLT3 mutations (median age, 49 years; range 5–68 years) were randomised to receive either oral lestaurtinib or placebo, for up to 28 days after each of the four courses of chemotherapy.95 Recently published data showed that lestaurtinib yielded no improvements in 5-year RFS and OS when added to first-line chemotherapy.95 Nevertheless, subgroup analysis indicated improved OS and significantly reduced rates of relapse in lestaurtinib-treated patients who sustained >85% FLT3 inhibition as assessed by the plasma inhibitory activity assay.95 In addition, elevated FLT3 ligand had no impact on lestaurtinib plus chemotherapy treatment.95
Three multitargeted TKIs currently approved for other malignancies have demonstrated activity against FLT3: ponatinib, sunitinib and sorafenib. In a phase I study of 12 previously treated patients with AML (58% had FLT3- ITD), ponatinib gave an overall response rate (ORR) of 25%.96 Following safety concerns, ponatinib was temporarily removed from the market in 2013.97 Since December 2013 the FDA has granted ponatinib full approval for the treatment of adult patients with chronic phase, accelerated phase or blast phase chronic myeloid leukaemia (CML) or t(9;22)-positive acute lymphoblastic leukaemia (ALL) for whom no other TKI therapy is indicated; and for the treatment of adult patients with T315I-positive CML or T315I-positive and t(9;22)-positive ALL. The full approval and label update was based on a 48-month follow-up data from the pivotal phase II PACE clinical trial of ponatinib in heavily pretreated patients with resistant or intolerant CML or t(9;22)-positive ALL.98,99 Currently, a dose escalation study of ponatinib, alone and in combination with 5-azacytidine, in patients with FLT3-mutated AML is planned at the MD Anderson Center but not yet recruiting.100 In addition, sunitinib in combination with intensive chemotherapy (cytosine arabinoside/daunorubicin induction followed by three cycles of intermediate-dose cytosine arabinoside) as maintenance therapy for 2 years showed promising findings in a phase I/ II trial of 22 AML patients with activating FLT3 mutations.101
Sorafenib demonstrated efficacy in phase I studies, and no dose-limiting toxicity was observed.102,103 The addition of sorafenib to chemotherapy has also yielded positive data in a phase I and II study.104,105 In the phase II study in younger adult (age range, 18–60 years) AML patients (n=267, of whom n=46 were positive for FLT3-ITD), median EFS was 9 months in the placebo group as compared to 21 months in the sorafenib group, corresponding to a 3-year EFS of 22% in the placebo group as compared to 40% in the sorafenib group (HR 0.64, 95% confidence interval [CI]; 0.45–0.91; p=0.013).104 In the subgroup analysis of FLT3-ITD positive AML, RFS (18 versus 6 months) and OS (not reached versus 19 months) were higher in the sorafenib group as compared to the placebo group.
The results in elderly AML patients with sorafenib in combination with standard intensive chemotherapy are controversial. Whereas one randomised double-blinded study in 197 elderly AML patients found no beneficial effect of the addition of sorafenib as compared to placebo,106 the opposite was the case in a recently published trial.107 Within this study, sorafenib was added to daunorubicin and cytarabine-based induction and consolidation chemotherapy and was also continued for 12 months of maintenance therapy. Fifty-four patients with a median age of 67 years (range, 60–83 years) were enrolled (n=39 were FLT3-ITD mutated (71%) and n=15 were FLT3-TKD (29%) mutated). The observed 1-year OS was 62% (95%-CI, 45–78%) for the FLT3-ITD patients (meeting the primary end point 62% versus 30% for a historical control group, p<0.0001) and 71% (95%-CI, 42–92%) for the FLT3-TKD patients. Nevertheless, the study by Serve et al.106 might have been biased, since the trial was not selected for the target population and the proportion of FLT3-ITD was very low in the study cohort (28 of 197 patients; 14%). In a phase II study of previously treated patients with AML (n=37, FLT3-ITD in 93%), a lower intensity regime with azacytidine yielded promising results: an ORR for response of 46% including incomplete count recovery (CRi) in 27%, CR in 16% and partial response (PR) in 3%. The median time to achieve CR/CRi was two cycles and the median duration of CR/CRi was 2.3 months.108 These findings suggest that further investigation of sunitinib and sorafenib in this treatment setting is warranted.
Midostaurin is currently the only TKI that has demonstrated convincingly superior results as compared to standard intensive therapy in younger FLT3-mutated AML patients for all survival end points including OS.109 Midostaurin affects multiple targets including c-Kit, platelet-derived growth factor receptors (PDGFR), as well as FLT3.110 In a phase I–II trial midostaurin 50 mg orally 2×/day given for 14 days was safely combined with standard induction therapy of daunorubicin and cytarabine in patients with newly diagnosed AML and a CR-rate of 80% could be achieved.111 These encouraging results provided rationale to move on to a randomised
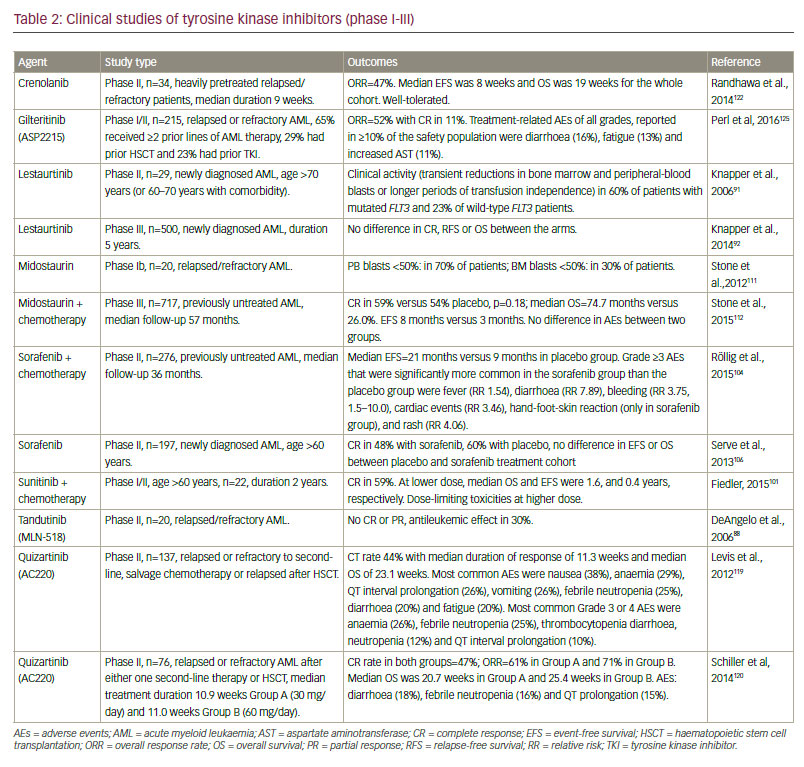
phase III trial CALGB 10603 (RATIFY; NCT00651261). The trial was activated in May 2008 and recruitment of over 700 younger adult FLT3- mutated, including FLT3-ITD and FLT3-TKD, AML patients (18–59 years) was finally achieved in October 2011. The study scheme consisted of the addition of midostaurin or placebo to standard intensive “7+3” induction chemotherapy as well as four cycles of high-dose cytarabine (HiDAC) as consolidation therapy. In all patients, maintenance therapy of 1 year with midostaurin or placebo according to initial randomisation was intended. Although not specifically mandated, allo HSCT was performed in 57% of the overall study cohort including transplants in refractory and relapsed patients. The combination of midostaurin to intensive chemotherapy significantly improved OS in younger adults with FLT3-mutated AML with a HR of 0.77 (95%-CI: 0.63–0.95, p=0.008), translating into a median OS of 74.7 months for the midostaurin arm (range, 31.7 months – not reached) as compared to 25.6 months for the placebo-arm (range, 18.6–42.9 months), respectively. Interestingly, this improvement was regardless of the FLT3 mutational status (either ITD or TKD) or the FLT3-ITD allelic ratio.112 Based on these results, on April 28, 2017, the US Food and Drug Administration (FDA) approved midostaurin (Rydapt®; Novartis; Basel, Switzerland) for the treatment of AML in newly diagnosed patients who are FLT3-mutation-positive as detected by an FDA-approved test, in combination with chemotherapy.113 In Europe, the marketing authorisation application for midostaurin is still under review by the European Medicines Agency (EMA). Based on a phase II followup study of the RATIFY trial in AML patients (age 18–70 years) with FLT3- ITD evaluating midostaurin in combination with intensive induction-, consolidation- including allo HSCT and maintenance therapy in all patients, the approval may be extended to older patients aged between 60 and 70 years.114
In addition, the combination of sequential azacitidine (intravenous 75 mg/m2 daily for 7 days) and escalating doses of oral midostaurin (25, 50 and 75 mg bid) on days 8–21 of a 28-day cycle has been investigated in a phase I study in untreated, elderly (median age: 73, range 57–83 years) and/or relapsed AML patients. No doselimiting toxicities occurred. Seventeen patients were enrolled and 14 patients were evaluable for response: three attained a CR and two had haematologic improvement. Median survival from enrolment was 6 months (range, 1 to =19 months). Interestingly, none of the patients harboured an FLT3 mutation. The authors concluded that the combination of sequential azacitidine and midostaurin was safe and tolerable with response rates comparable with azacitidine alone.115
The combination therapy of midostaurin and azacitidine was also evaluated in a phase I/II study (n=54; 74% had a FLT3 mutation; 76% had been previously treated). During the dose-finding part of the study, six patients received midostaurin at a dose of 25 mg bid and eight at a dose of 50 mg bid. No dose-limiting toxicities occurred. Among the 54 patients in the phase II study, after a median of 12 weeks (range, 1–31), the ORR was 26%. One patient (2%) achieved a CR, six (11%) achieved a CR with CRi, six (11%) a morphologic leukaemia-free status (defined as <5% blasts in the bone marrow regardless of neutrophil and platelet count in the peripheral blood) and one patient (2%) a PR.115,116 Nevertheless, even with the addition of midostaurin to intensive therapy including allo HSCT and maintenance therapy within the RATIFY trial, a significant proportion of the patients still relapsed within the first 2 years,112 raising the question as to whether or not more selective TKIs would be more beneficial.
Second-generation FLT3 TKIs including quizartinib, crenolanib, PLX3397 and gilteritinib (formerly ASP2215), are more potent and selective based on cell cultures and animal models than the first-generation inhibitors.117 Quizartinib, a novel bis-aryl urea, is very specific for FLT3, has a high capacity for sustained FLT3 inhibition and an acceptable toxicity profile.118 In a phase II study quizartinib demonstrated particular efficacy in patients with FLT3- ITD mutations (n=137) who were relapsed or refractory to second-line, salvage chemotherapy or relapsed after allo HSCT.119 The CR rate was 44% with a median duration of response of 11.3 weeks and median OS of 23.1 weeks. Of note, one-third of patients were successfully bridged to allo HSCT. A subsequent phase II study recruited 76 patients with FLT3-ITD mutations, with relapsed or refractory AML after either one second-line therapy or allo HSCT. Patients were randomised to quizartinib 30 mg/day (Group A) or 60 mg/day (Group B) given orally during 28-day continuous treatment cycles, until relapse, intolerance or proceeding to allo HSCT. The ORR was 61% in Group A and 71% in Group B. In addition, 32% of patients in Group A and 42% in Group B could be successfully bridged to allo HSCT.120 A phase III study of quizartinib or placebo with induction and consolidation chemotherapy, and as maintenance in patients with newly diagnosed FLT3- ITD AML, is ongoing (age range: 18–75 years, planned inclusion number: n=536; ClincalTrials.gov identifier: NCT02668653).121 However, resistance to quizartinib in FLT3-TD has been reported; this has been attributed to acquired D835Y TKD mutation on the FLT3-ITD allele.57
Other second-generation FLT3 inhibitors have also yielded positive findings. Gilteritinib and crenolanib are able to inhibit both FLT3-ITD and FLT3-TKD mutations. High response rates have been reported in two clinical studies of crenolanib, particularly in FLT3 inhibitor-naive patients (phase II).122,123 In a single centre phase II study (n=34), patients had received a median of 3.5 prior therapies (sorafenib in 57%, quizartinib in 14%, PLX3397 in 5% and midostaurin in 10%; 9% and 5% had received two and three FLT3 TKIs, respectively). At a median follow-up of 14 weeks, the ORR was 47%: 12% achieved CRi, and 3% morphologic leukaemia-free state. The median EFS was 8 weeks and OS was 19 weeks for the whole cohort.122 In another phase II study of relapsed/refractory patients (n=19, median age 47 years), one patient had a CR while two had a CRi and four patients were bridged to transplant.123 These preliminary data suggest that crenolanib is very promising in relapsed and refractory AML patients, and further trials are being initiated (e.g., NCT02298166, NCT02400281, NCT02270788, NCT02283177).
In a phase I/II study (n=252) of heavily pretreated patients (70% had =2 prior AML therapies, 29% had prior HSCT, and 25% had prior TKI treatment, most commonly sorafenib) receiving gilteritinib, FLT3-ITD patients showed an ORR of 52%, with CR in 11%. Clinical responses occurred in FLT3-mutated patients with ITD, D835 and both mutations (ORR: 55%, 17% and 62%, respectively). The median OS for FLT3-mutated patients receiving gilteritinib =80 mg was around 31 weeks.124 A phase III trial of gilteritinib is currently ongoing (NCT02421939, estimated enrolment 369). Patients with FLT3-mutated AML in first relapse or refractory to frontline therapy are being recruited and randomised to treatment with either gilteritinib or to investigator’s prerandomisation choice of specified salvage chemotherapy. The primary objective is OS; key secondary objectives are EFS and CR rate.125
TKI treatment postallogeneic HSCT
In addition, the efficacy of TKIs following allo HSCT is being investigated. A retrospective multicentre study of 29 patients who had undergone allo HSCT, treatment with sorafenib led to haematological remission in 37%, bone marrow remission in 8%, CR (with and without normalisation of peripheral blood counts) in 23% and molecular remission with undetectable FLT3-ITD mRNA in 15%, respectively. Allo HSCT patients developed sorafenib resistance less frequently (38% versus 47%) and significantly later (197 days versus 136 days, p=0.03) than those without prior HSCT, and sustained remissions were seen only in the allo HSCT cohort.102 The addition of midostaurin to intensive induction therapy and as maintenance after allo HSCT or HiDAC is currently being investigated. Preliminary data indicate that this approach was feasible and outcomes were favourable compared with historical data, particularly in patients with a high FLT3-ITD mutant to wild type ratio.114 An ongoing trial is also investigating the efficacy and safety of quizartinib, postallogeneic transplant (NCT02668653).121
Mechanisms of resistance
Patients who relapse after treatment with a TKI can develop point mutations in the target kinase domain as a mechanism of resistance.57,126 Resistance has also been associated with upregulation of parallel and downstream signal transduction pathways, and may also involve stromal cells of the bone marrow.127 In addition, an interaction between CD34+ progenitor cells from patients with FLT3-ITD mutations and niche cells has been reported in another publication.128 This interaction enables the maintenance of leukemic progenitors in the presence of a TKI.128
Different FLT3 TKIs display distinct and nonoverlapping resistance profiles in vitro; TKD1 mutations showed a response to SU5614, sorafenib and sunitinib but diminished response to PKC412, whereas TKD2 mutations were sensitive to PKC412, sunitinib or sorafenib.56
Another mechanism of resistance might be related to the FLT3-ITD insertion site.129 These data suggest that combinations of FLT3 inhibitors may be required to prevent FLT3 resistance mutations in FLT3-ITDpositive AML. Some research has suggested that FLT3 inhibitor therapy combined with crenolanib may prevent the emergence of resistance.49
Future developments
Targeting multiple pathways may be necessary to ensure enduring responses, leading to a focus on combined treatment regimens. A phase I clinical trial evaluating the combination of the mammalian target of rapamycin (mTOR)-inhibitor RAD001 with midostaurin is ongoing.130 Homoharringtonine has been shown to act synergistically with FLT3 inhibitors.131 In addition, preclinical data suggest that a number of PI3K, AKT, mTOR and MEK inhibitors may act synergistically with FLT3 inhibition.132–134 Recently, dual inhibition of FLT3 and Pim kinases has been found to eradicate FLT3-ITD mutated AML cells in vitro.135 However, concern has been expressed that targeting multiple pathways may result in increased toxicity; therefore, more clinical data are needed on these combinations.
Other multitargeted TKIs are also in early stage clinical development. Pacritinib (formerly SB1518) is a TKI with activity against FLT3 and Janus kinase 2.136 The first clinical study of pacritinib showed promising results.137
Summary and concluding remarks
Increased understanding of FLT3 mutations in AML has presented an opportunity for the use of targeted therapies, and thus broadened a treatment landscape that has remained unchanged for decades. The incorporation of FLT3 TKIs into current treatment paradigms should lead to a significant improvement in the prognosis for AML patients with activating FLT3 mutations. Of the several promising therapeutic agents that are in clinical development, midostaurin is at the most advanced stage and is the first targeted agent to improve survival in AML with FLT3 mutations in combination with intensive chemotherapy and/or allo HSCT including maintenance therapy in younger, adult AML with FLT3- mutations. Since midostaurin was FDA approved on April 28, 2017, its use according to the CALGB 10603/RATIFY trial to treat younger adult AML patients with FLT3-mutations seems currently to be the best approach for this patient group. Whether newer, more selective TKIs might be clinically more beneficial is currently being tested in clinical trials.
The optimum use of FLT3 TKIs remains an active area of research. Numerous questions remain unanswered, including the optimal sequencing of second-generation FLT3-specific agents and multikinase TKIs. Complete and sustained inhibition of FLT3-mutated AML may require a combination of agents, both targeted and conventional chemotherapy, but at present the optimal schedule is not known. The optimum role of FLT3 TKIs in relapsed/refractory patients as a bridge to allo HSCT or as post-HSCT maintenance remains to be established. There is a need for further randomised clinical trials to investigate these questions. Further research will expand our understanding of FLT3 mutations and the mechanisms of resistance to FLT3 TKIs.