
The coagulation cascade
The formation of a blood clot inside a blood vessel obstructs the blood flow through the circulatory system. Hemostasis is the process that prevents blood loss after any injury. The coagulation process that directs hemostasis constitutes a complex set of reactions.
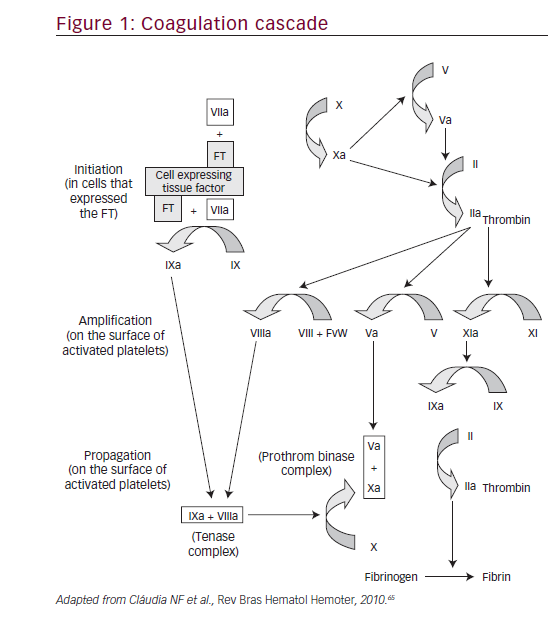
In a study conducted by Davie and Ratnoff (1964),1a simple waterfall sequence is proposed to explain the function of the various protein clotting factors during the formation of the fibrin clot. When clotting is initiated, fibrinogen is converted to a form that exhibits enzymatic activity, and this occurs in a stepwise sequence where each newly formed enzyme reacts with its specific substrate, thus converting it to an active enzyme. The concept of the coagulation cascade as a series of stepwise enzymatic conversations was first proposed by MacFarlane (1964).2According to MacFarlane, there exist two pathways: the extrinsic pathway, involving tissue factor and factor VII, and the intrinsic pathway, involving factors such as XII, XI, IX, VIII, and V. Both pathways converge to activate factor X and lead to the transformation of prothrombin into thrombin. Subsequently, the fibrinogen gets converted into fibrin by the action of thrombin. Within this cascade model, the role of platelets in coagulation was considered as an independent mechanism.3During the following three decades, many studies were undertaken, culminating in simultaneous publications from two groups located in Houston and North Carolina.4,5Both groups described a ‘new cascade’, and this has been internationally accepted and demonstrated by a recent publication.6 This new perspective built on the classical cascade has been represented in the following ways.
• The complex formed by tissue factor and factor VII contributes to the activation of factor IX (FIX), showing that the intrinsic and extrinsic coagulation pathways are linked almost from the beginning of the process.
• The complete process does not occur continuously, but rather in three consecutive phases: an initial phase, an amplification phase, and a propagation phase. The platelets and thrombin are actively involved in the amplification and propagation phases.3
Coagulation is a complex process in which circulating cells and coagulation factors interface with tissue-based proteins to form an insoluble clot at the sites of vascular injury. The coagulation process involves a complex set of reactions involving approximately 30 different proteins.7These reactions
convert soluble fibrinogen into insoluble strands of fibrin, which, together with platelets, forms a stable thrombus. Even though this dynamic process represents an advantageous response after localized vessel injury, clot formation may also be undesirable. For instance, thrombosis within the coronary beds is the proximate cause of myocardial infarction. Therefore, the development of pharmacologic agents that attenuate safe and effective clot formation is an attractive goal for clinicians and the pharmaceutical industry.8
The initial step in the pathogenesis of cardiovascular diseases, kidney failure, stroke, infectious diseases, and cancer, is endothelial dysfunction. Vascular endothelial cells line the entire circulatory system. These cells have very unique and distinct functions including fluid filtration, hormone trafficking, blood vessel tone, and hemostasis. One of the key functions of endothelial cells is the inhibition of non-physiologic initiation of blood coagulation, resulting in thrombosis.9 Numerous coagulation cascade models have been proposed, including the intrinsic and extrinsic pathway models and the more recent cell-based model.
Intrinsic and extrinsic pathway models
The intrinsic and extrinsic pathway models divide the initiation of coagulation into two distinct parts.10
The extrinsic pathway might be responsible for the initial generation of activated factor X (factor Xa), whereas the intrinsic pathway leads to the amplification of factor Xa generation. Factor Xa plays a central role in the coagulation cascade because it occupies a point where the intrinsic and extrinsic pathways converge.10
Cell-based model of coagulation
Coagulation in vivo is best characterized as a harmonized series of cellbased events. This model represents the interaction between cellular activity and coagulation proteins that leads to thrombus formation and hemostasis.11 Replacing the conventional ‘cascade’ hypothesis, the cell-based model of hemostasis proposes that coagulation takes place on different cell surfaces in four distinct steps: initiation, amplification, propagation, and termination (Figure 1).
Factor IX
FIX plays a key role in blood coagulation because deficiency or absence of its activity results in an X-linked bleeding diathesis, hemophilia B.12As mentioned earlier, thrombin may activate FXI to FXIa on the platelet surface by a feedback mechanism to allow additional activation of FIX to FIXa for sustained and consolidated coagulation.13In the present scheme of coagulation, activation of FIX by tissue factor (TF)/VIIa and FX by FIXa is significant because initial FXa generated by TF/FVIIa complex combines with TF pathway inhibitor (TFPI), and the resultant FXa/TFPI shuts down TF/FVIIa activity.14,15Therefore, the FXa produced by the FIXa/FVIIIa complex represents a crucial step in coagulation, and disruption of this step may represent a possible target for the development of new antithrombotics.
Factor IX structure
FIX is a vitamin K-dependent protein that is synthesized by hepatocytes as a precursor of a serine protease, FIXa. The gene for FIX consists of eight exons and seven introns, which is approximately 34 kb long, and is located on the long arm of the X chromosome at Xq27.1.16It is synthesized as a precursor protein of 461 amino acids containing a 28-residue signal prepeptide and an 18-residue leader propeptide.
Factor IX-platelet interface
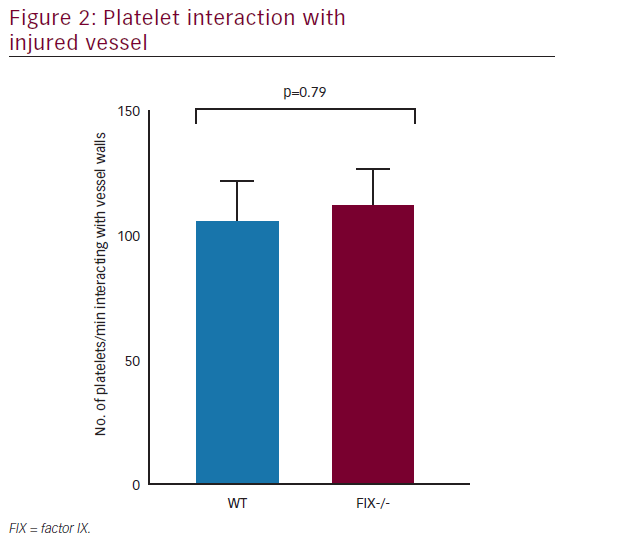
The contribution of platelets to hemostasis and thrombosis is well explained; yet, rising evidence supports the presence of numerous distinct platelet populations within a developing thrombus—each with very specific functional roles.17FIXa, viewed originally as a protein responsible exclusively for clot formation, also plays a primary role in platelet-mediated hemostasis as well(Figure 2).
The binding of FIX and FIXa to thrombin-activated human platelets is well described, with 300–400 sites per platelet.18The presence of FVIIIa and FXa increases the affinity of receptors for FIXa that in turn participates in FX activation. Although hemostasis and thrombosis have different implications, a considerable overlap is noted in the molecular mechanisms involved in these interactions resulting from a coordinated series of events such as adhesion and promotion of coagulation.
Venous and arterial thrombosis
Thrombosis is the leading cause of morbidity and mortality worldwide.19Arterial thrombosis is the most common underlying cause of acute myocardial infarction, peripheral vascular disease, and non-hemorrhagic cerebrovascular accidents. Venous thrombosis is considered to be a multicausal disorder. Multiple genetic and environmental factors contribute to the development of the disease. Most of these factors relate to changes in the composition of the blood and blood flow. In combination with a damaged endothelium, they create a hypercoagulable state locally. When the hypercoagulability surpasses a certain threshold, excessive thrombus formation will occur, which may present as thrombosis of the large veins of the arms and legs (deep vein thrombosis [DVT]), superficial veins (STP), and pulmonary embolism (PE).20While arterial thromboembolic events are the foremost cause of death and disability, the acute manifestations of venous disease can cause serious disorders.21
Venous and arterial thrombosis have traditionally been regarded as separate diseases, each with their own pathophysiologic basis, unique risk factors, and distinct therapeutic regimens. The consequences of arterial thrombosis, such as myocardial infarction and stroke, are the most common causes of morbidity and mortality globally22Though the clinical manifestation of myocardial infarction and strokes are different, they are the result of the same pathogenic process, i.e. formation of a thrombus over an underlying atherosclerotic plaque in the setting of high blood flow and high shear arterial circulation.23Arterial clots typically occur in an injured vessel and the most common cause of vascular damage in the arterial system is atherosclerotic vascular disease (ASVD).21The risk factors for arterial thrombosis are, therefore, considered the same as those for ASVD. While vascular injury can promote the formation of venous clots, stasis and changes in blood components (thrombophilia) are the most important risk factors for venous clot development.21 Venous clots occur in a low flow system; they are rich in fibrin that is enmeshed with red blood cells and are referred to as red clots.20,24
Recent epidemiologic studies have documented an association between these vascular complications, probably due to the presence of more overlapping risk factors than were previously recognized.25Risk factors such as age, obesity, infections, and metabolic syndrome have been found in both conditions. The existence of an association is further supported by the finding that patients with venous thromboembolism are at risk of arterial events and vice versa.26
Elevated plasma coagulation factor levels
The risk of thrombosis increased linearly with plasma factor level (FVII, FIX, FXII, and FII). For others, thrombosis risk was only associated with levels above the 90th percentile (FVII, FV, FX, TAFI) or below the 10th percentile (TFPI) of the distribution in the normal population.27
Factor IX and thrombosis
As mentioned earlier, FIX plays a key role in blood coagulation, as shown by the bleeding tendency associated with congenital FIX deficiency (hemophilia B, Christmas disease). The latest studies show that, after activation of FIX by the tissue factor/FVIIa complex, or by FXIa, FIX plays a key role in thrombin generation in the vicinity of platelets and that FIXa is the thrombogenic trigger after infusion of prothrombin complex concentrates (PCC).28It plays a major role in wound healing, vascular repair, and angiogenesis. Recent case-control studies have shown that high levels of FIX are associated with increased risk of venous thromboembolism. Moreover, epidemiologic studies in large random population samples have revealed that FIX levels are associated with several thrombotic risk factors, including age, obesity, oral estrogen use, smoking, blood pressure, and low social class. Furthermore, FIXa is also increased in patients with acute coronary artery thrombosis.29
Factor IX and venous thrombosis
Increase in plasma levels of several coagulation factors such as FVIIIc, fibrinogen, FXIc and FXIII have recently been associated with the risk of DVT.30–34
Van Hylckama Vlieg et al.35studied the relationship of FIX antigen with DVT in the Leiden Thrombophilia Study (LETS). Subjects with plasma IX antigen above the 90th percentile (≥129 IU/dI) had a 2.5 (95% confidence interval [CI] 1.6, 3.9) increased the risk of DVT. The risk factors appeared to be higher in females than in males and they were higher in premenopausal females not using oral contraceptives. Lowe et al. also studied the relationship of FIX to DVT and showed that an increased level of FIX is associated with venous thrombosis.29
Thus, the results of these studies suggest that high FIX (activity or antigen) may be a mechanism for venous thrombogenesis. High levels of FIX might be genetic or reflect environmental effects of risk factors such as estrogen, age, blood lipids, or obesity. Additional studies are essential to establish the relationships between FIX, risk factors, and venous thromboembolism.
Factor IX and arterial thrombosis
Patients with hemophilia A or B have a lower risk of coronary heart disease (CHD) than the general male population.36,37The plasma levels of FIX activity (FIXc) are associated not only with FVIII activity (FVIIIc) but also with several CHD risk factors in the general population.38,39The association of FIXc with CHD risk factors were highest in both women and men for triglycerides,39 which may be of significance given the ability of triglyceride-rich very-low-density lipoprotein (VLDL) and other lipoproteins to bind vitamin K-dependent coagulation factors (including FIX) and support procoagulation enzymatic complexes in thrombin formation.40,41However, there are no reported prospective cohort studies of FIX and risk of stroke or CHD.
Myocardial infarction has occasionally followed infusion of PCCs.42,43The importance of FIXa in PCC for thrombosis has been noted.44 In acute myocardial infarction or acute unstable angina pectoris (acute coronary syndromes), plaque rupture exposes circulating blood to tissue factor FVIIa complexes, which can activate factors IX and XI on the local platelet/lipid surface. Minnema et al.45 reported increased plasma levels of FIX activation peptides in patients with acute coronary syndromes, compared with patients with stable angina. This paper provides the first evidence for FIX activation in acute coronary syndromes. Such activation may play a key role in coronary thrombogenesis through the continuous generation of thrombin and fibrin formation, in addition to the inhibition of endogenous fibrinolysis through activation of TAFI.46
Antithrombotic therapy and factor IX
Oral anticoagulants decrease the activity of vitamin K-dependent coagulation factors including prothrombin, FVII, FIX, and FX. There has been recent interest in the increased sensitivity of some patients to oral anticoagulant-induced decrease in FIXc used routinely for monitoring of the oral anticoagulant effect. Such patients may have anti-phospholipid antibodies or mutations in the propeptide of FIX, causing a reduced affinity of the carboxylase for FIX precursor.47–49 Patients with these mutations have normal baseline FIXc levels, resulting in severe bleeding and a markedly prolonged activated partial thromboplastin time (APTT). In a large study, Legnani et al.49 observed that FIX levels varied greatly regardless of the similar achieved anticoagulation intensity, making it difficult to identify those with very low FIXc levels from the APTT. The group provided a table of ranges for FIXc and APTT for INR classes that may help in such identification.
Heparins also lower the levels of FIXc, which contributes to their anti-thrombotic effects.50 Due to the potential importance of FIXa in thrombogenesis in animal models in vitro, in idiopathic venous thrombosis, in PCC-induced thrombosis and coronary thrombosis, it shows suitability to study selective FIX inhibitors as antithrombotic therapy.44,51–53 FIXa can be inhibited by various mechanisms such as chemically, by an antibody, or by blocking of the active site.54,55
Furthermore, FIXa inhibitors may have a higher ratio of anti-thrombotic activity to bleeding risk that heparins have in animal models.55–59Two studies have proposed that the main effect of heparin on the prolongation of the APTT is through blocking of the FIXa activation of FX.60,61 Studies in baboons and dogs revealed that when active site-inhibited FIXa is used as an anticoagulant in place of heparin, the extracorporeal circuit remains free of fibrin deposition, and intraoperative blood loss is significantly diminished relative to the standard heparin protocol.62,63 Furthermore, an antibody directed against the amino-terminal region of FIX acts as a powerful antithrombotic agent in rat and guinea pig models of arterial thrombosis.64
Pegnivacogin is also an anti-FIX aptamer that inhibits coagulation FIX activation, preventing FV-mediated generation of thrombin. While the drug is on hold pending further studies, it has been shown to rapidly induce anticoagulation, thus providing safe and stable anticoagulation in the preclinical and clinical settings.
New oral anticoagulants that inhibit either FIXa (rivaroxaban, apixaban) or thrombin (dabigatran etexilate) have gained approval in many countries owing to some clinical indications. Future clinical experiments of FIXa inhibitors in prophylaxis and treatment of thrombosis might test the theory that high levels of FIXa play a key role in venous and arterial thrombogenesis.
Conclusion
In conclusion, it is important to shed light on the importance of FIXa, as it is a key intermediary in the intrinsic pathway and targeted inhibition of FIXa-dependent coagulation might inhibit microvascular thrombosis development. Further studies of the theoretical benefits of targeting FIXa compared with former anticoagulant targets will translate into better outcomes.











