In recent years, our understanding of the complement system and its role in disease has increased dramatically. With the advent of complement 5 (C5) and C3 inhibitors, our ability to manipulate the complement system has improved the outcomes and quality of life for patients with paroxysmal nocturnal haemoglobinuria and atypical haemolytic–uraemic syndrome.1–3 More recently, this understanding has been applied to cold agglutinin disease (CAD).4 Because of the increased risk of infection with complement inhibition, blockade at the C1s level is being explored to strategically inhibit the complement system while minimizing the infection risks.5
Sutimlimab is a humanized immunoglobulin (Ig) G4 antibody developed to inhibit the classical complement pathway.6,7 It targets C1s, the serine protease of the C1 complex, which activates the classical complement pathway.8 By blocking the initial stages of complement, the generation of C3 convertase by the classical pathway is diminished, reducing C3b membrane deposition and subsequent terminal complement activation.9 However, because the lectin and alternative pathways remain intact, this may reduce the risk of infection associated with complement blockade.6–8
Cold agglutinin disease
CAD is a very rare haemolytic anaemia characterized by antibody-mediated haemolysis, complement activation, thrombosis and poor quality of life.6,7 Around 15–25% of autoimmune haemolytic anaemias are associated with IgM ‘cold’ antibodies.10–13 Primary CAD is characterized by an underlying, non-malignant, B-cell lymphoproliferation, resulting in complement-mediated haemolysis via the classical complement pathway.10–13 In contrast, secondary cold agglutinin syndrome is defined by cold agglutinin-induced haemolysis associated with an underlying infection, such as Mycoplasma pneumonia or Epstein–Barr virus, or overt lymphoid malignancy.11
In contrast to ‘warm’ autoimmune haemolytic anaemia, where the antibodies are mostly IgGs, the antibodies in CAD are IgMs.10,14 IgMs may be clonal or polyclonal, with a predominance of kappa chains.11 Antibodies in patients with primary CAD are preferentially encoded by the immunoglobulin heavy variable (IGHV)4-34 gene segment.14–16 These cold antibodies interact with the red blood cell (RBC) membrane antigen via weak hydrogen (van der Waals) bonding and are strongest at 4–30°C.14,17 The thermal amplitude of the antibody is defined as the temperature or temperature range at which the maximal antibody binding to the antigen will occur. Because IgM antibodies are pentamers, they can bind to multiple RBCs, causing agglutination.17 Cold-induced circulatory symptoms such as acrocyanosis and Raynaud’s are due to RBC agglutination.14,17,18 However, in addition to inducing agglutination, IgM molecules always bind the RBC membrane complement and activate the complement cascade.14 For intravascular haemolysis to occur, a lot of complement needs to bind to the RBC.
Regardless of the aetiology, most cold agglutinins are IgM antibodies capable of complement fixation to the RBC membrane.18 In secondary cold autoimmune haemolytic anaemia, most antibodies are directed to the ‘I’ antigen, with the exception of the cold agglutinin syndrome associated with Epstein–Barr virus, where the antibody is directed to the ‘i’ antigen.11 In syphilis and some other viral syndromes, the antibody is an IgG antibody, which binds to the ‘P’ antigen.19 This antibody, known as the Donath–Landsteiner antibody, has a biphasic thermal amplitude that allows it to bind to the RBC membrane at <20°C and fix complement.19 This complement-fixing IgG antibody causes both extravascular and intravascular haemolysis (paroxysmal cold haemolytic anaemia; Figure 1).11,19, 20
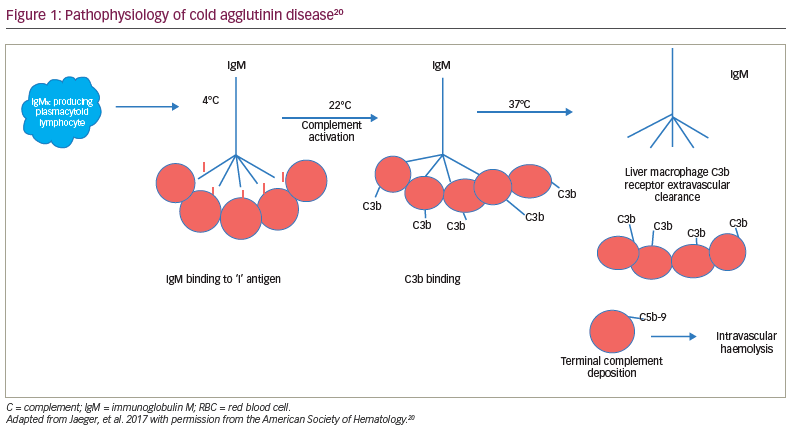
Due to the IgM antibodies, the complement system is activated through the classical complement pathway.17 The ‘I/I’–IgM complex binds to C1, thereby activating the serine proteases C1r and C1s.18 This leads to the activation of the ‘C1 qrs’ complex and the creation of C4b/2a, or C3 convertase. This results in C3 cleaving to C3a, an inflammatory protein, and C3b, which binds to the membrane.21–23 The downstream consequences are the extravascular opsonization of the C3b-coated RBCs, inflammation and thromboembolic complications. Intravascular haemolysis may occur with the subsequent cleavage of C5, leading to the deposition of the membrane attack complex, C5b-9.21–23 However, anaemia in CAD is primarily due to the extravascular hemolysis macrophage clearance of C3b-coated RBCs in the liver, which has a receptor for C3b.20–23 This opsonization of RBCs by C3b appears to be the main cause for RBC destruction in CAD and, therefore, may be a target for treatment. Blocking the classical complement pathway is a logical strategy for improving outcomes in CAD.18–23
Patients with CAD have poor quality of life due to anaemia but also cytokine-induced inflammation and thromboembolic complications.11,24,25 Inhibiting the classical complement pathway may reduce inflammation and thromboembolic events in patients with CAD.24,25 Data suggest that inhibiting C1 decreases inflammatory cytokines, interleukin (IL)-6 and tissue factor mRNA expression.26 This has been demonstrated in a clinical trial, with durable decreases of key cytokines IL-6 and IL-10 with C1s inhibition, suggesting that inflammation may play an important role in CAD.25 Furthermore, decreases in both IL-6 and IL-10 levels were associated with improvements in fatigue, suggesting that complement-mediated chronic inflammation may contribute to the non-haematological manifestation of fatigue in patients with CAD.25
There are no approved therapies for CAD. Off-label use of CD20-expressing B-cell-depleting therapy (rituximab monotherapy) induces a partial response in around 50% of patients, with a median response delay of 1.5 months and relapses occurring within 1 year.27 Adding cytotoxic agents, such as bendamustine or fludarabine, to rituximab was associated with improved response rates but more serious toxicity, including severe neutropenia.28,29
Since the classical complement pathway plays such an important role in CAD, C1s inhibition has been evaluated in several pivotal trials. Initial reports demonstrated markedly reduced haemolysis and RBC opsonization with TNT003, an IgG2a mouse monoclonal antibody that targets C1s, the classical-pathway-specific serine protease.30 In ex vivo haemolysis assays with 40 unique plasma samples from patients with CAD, TNT003 prevented haemolysis and anaphylatoxin generation driven by complement activation.30 As well, plasma samples contained reduced levels of upstream classical pathway components but not C5 (terminal complement), suggesting that the classical complement pathway is activated before the terminal cascade.30 Based on these preliminary data, sutimlimab (formerly BIVV009), a highly humanized IgG4 antibody to C1s, was developed and is being evaluated in clinical CAD trials.6,7
A phase Ib trial in patients with CAD demonstrated a rapid increase in haemoglobin level (median 3.9 g/dL at 6 weeks) with sutimlimab treatment, showing proof of concept.31 Patient follow-up indicated durable long-term benefits. The minimally effective dose for a biweekly infusion regimen was initially chosen as 45 mg/kg and then increased to 60 mg/kg for the pivotal trials CARDINAL and CADENZA. The CARDINAL trial was an open-label, single-arm, phase III trial that evaluated sutimlimab in patients with CAD with a recent history of RBC transfusion.6 The CADENZA trial was a randomized, phase III, double-blind, placebo-controlled trial that evaluated sutimlimab in patients with CAD with haemoglobin levels ≤10 g/dL but without recent transfusion.7 Both trials had a 26-week initial treatment period (Part A), followed by a long-term extension period (Part B).6,7 Their design and key results are summarized in Table 1.6,7,32
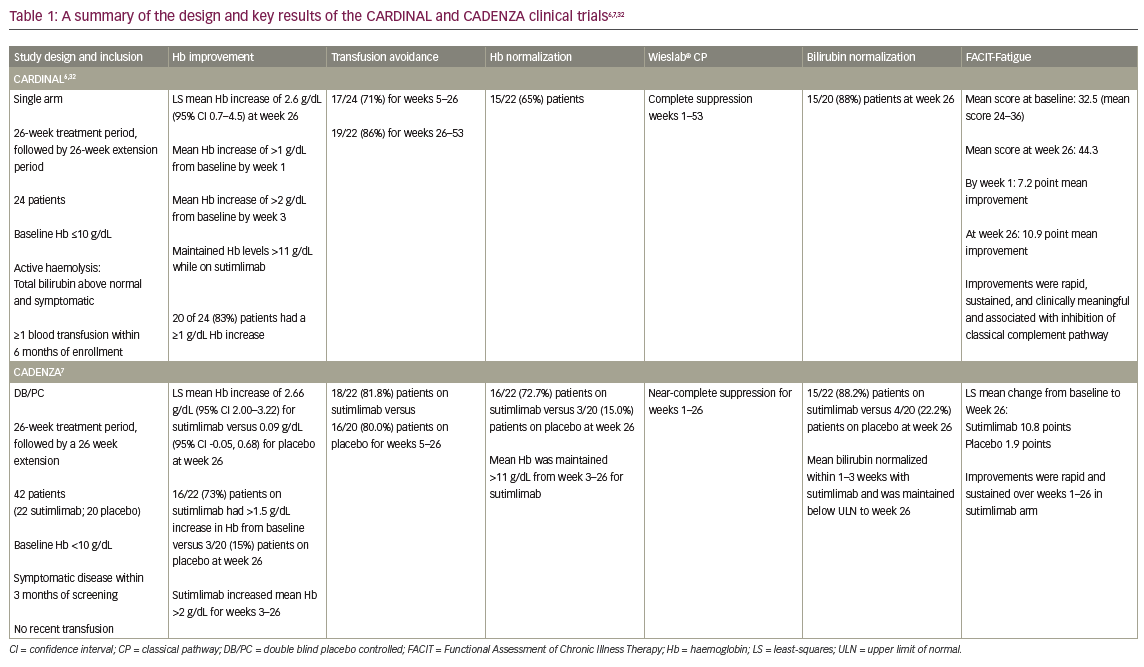
In the CARDINAL trial, the primary endpoint was a composite of normalization of haemoglobin to ≥12 g/dL or an increase in haemoglobin of ≥2 g/dL from baseline without requiring transfusion in weeks 5–26.6 With sutimlimab, 13 of the 24 enrolled patients (54%; 95% confidence interval [CI] 33–74) met the composite primary endpoint. Furthermore, the mean ± standard error (SE) haemoglobin levels improved rapidly from baseline by 1.2 ± 0.3 g/dL in the first week and 2.3 ± 0.3 g/dL by the third week (Figure 2).6,33 Mean haemoglobin levels were maintained at ≥11 g/dL for weeks 3–53.6,32 Of the 22 evaluable patients in Part B, 11 (55.0%) had normalized haemoglobin at week 53.32
Along with the improvement in haemoglobin, the mean total bilirubin rapidly decreased at week 1, normalized (defined as <20.5 μmol/L) by week 3 and remained normal to week 53 (Figure 2).6,32,33 Interim Part B data showed that 14/22 patients (63.6%) achieved normal bilirubin level at week 53.33 At week 53, the mean ± SE change in bilirubin from baseline was -35.3 ± 4.2 μmol/L.33 Moreover, among patients with available lactate dehydrogenase (LDH) data (n=21), the mean ± standard deviation LDH was 367.2 ± 253.6 at week 53.32
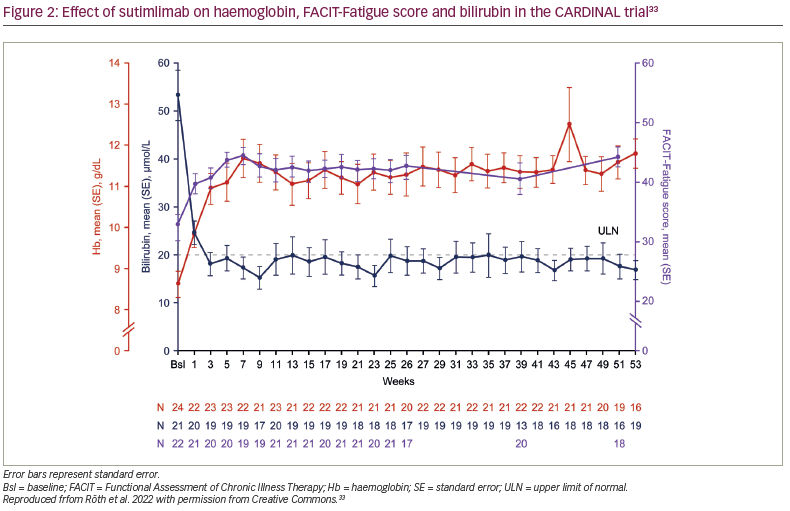
Sutimlimab treatment was associated with near-complete inhibition of classical complement pathway activity, which was measured by the Wieslab® Complement System Classical Pathway assay (Svar Life Science, Malmö, Sweden).6,33 Mean ± SE classical pathway activity at baseline was 20.0% ± 3.4%. After the first dose of sutimlimab, at week 1, it was reduced from baseline and was maintained at 3.0% ± 0.7% at week 25 and 3.0% ± 0.7% at week 51. Mean ± SE total C4 activity was 0.04 ± 0.02 g/L, 0.29 ± 0.02 g/L and 0.24 ± 0.11 g/L at baseline, week 25 and week 51, respectively, consistent with the classical pathway inhibition activity (Figure 2).33 Mean absolute reticulocyte count was reduced, alongside increased haptoglobin levels. The reticulocyte count decreased from baseline to week 26, supporting the reduction in haemolysis.6,32 Treatment with sutimlimab was associated with rapid and durable decreases in key cytokines (IL-6, IL-10) from baseline to week 26, highlighting the influence of complement inhibition on inflammation in CAD.25 Inverse changes over time were observed for these select inflammatory cytokines and the Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue scores (Figure 2).33 These results support the idea that, in addition to anaemia, complement-mediated chronic inflammation contributes to the fatigue experienced by patients with CAD.23,25,26
In the CADENZA trial, the composite primary endpoint was the percentage of patients achieving response at week 26, defined as having not received transfusion or treatment for CAD during weeks 5–26, and with a haemoglobin level increase of ≥1.5 g/dL from baseline.7 Of the 42 patients investigated (22 receiving sutimlimab, 20 placebo), the primary endpoint was met by 16 patients (72.7%; 95% CI 49.8–89.3) in the sutimlimab arm versus three patients (15.0%; 95% CI 3.2–37.9) in the placebo arm. Patients who received sutimlimab were significantly more likely to achieve response criteria than patients on placebo (odds ratio 15.9, 95% CI 2.9–88.0; p<0.001). By week 3, sutimlimab, but not placebo, had increased mean haemoglobin and FACIT-Fatigue scores through to week 26 and had normalized mean bilirubin in 15 (88.2%) of patients versus 4 (22.2%) of patients on placebo at week 26.7
As seen in the CARDINAL trial, sutimlimab almost completely suppressed the classical pathway complement activation as measured by the Wieslab assay and C4 levels in the CADENZA trial.7 Sutimlimab treatment was associated with near-complete inhibition of classical complement pathway activity at week 1, which was sustained to week 26. Mean ± SE pre-dose classical pathway activity at baseline was 22.4% ± 4.2% in the sutimlimab arm and 32.8% ± 6.4% in the placebo arm. After the first dose of sutimlimab at week 1, mean classical pathway activity was reduced from baseline and was maintained through the treatment assessment time at week 25. Unlike in the placebo arm, reductions in mean ± SE classical pathway activity were maintained to week 26 in the sutimlimab arm (sutimlimab, 3.2% ± 0.5% versus placebo, 29.3% ± 7.0%). Mean ± SE pre-dose total C4 activity in the sutimlimab arm was 0.06 ± 0.01 g/L, 0.24 ± 0.01 g/L and 0.31 ± 0.02 g/L at baseline, week 1 and week 26, respectively, consistent with the classical pathway inhibition activity. In contrast, mean ± SE C4 activity in the placebo arm was unchanged from baseline (0.07 ± 0.02 g/L) through week 26 (0.07 ± 0.02 g/L).
In the sutimlimab arm, improvements in mean haemoglobin levels to ≥11 g/dL were maintained for weeks 3–26. Absolute reticulocyte count and LDH decreased, and haptoglobin levels increased. These improvements coincided with normalized C4 levels and near-complete classical pathway inhibition. Mean C1q levels remained unaffected over the 26-week study period.7
In the CARDINAL trial, from baseline to week 53, all 24 patients experienced ≥1 treatment-emergent adverse event (TEAE); in Part A, nine patients (37.5%) had ≥1 TEAE related to sutimlimab (as assessed by the investigator) (Figure 3).32,33 Of the 281 TEAEs, the most common TEAEs (>10%) were diarrhoea and nasopharyngitis (20.8% each); anaemia, nausea and hypertension (16.7% each); and upper abdominal pain, confusional state, constipation, contusion, cough, cyanosis, cystitis, dizziness, fatigue, gastroenteritis, haemorrhoids and upper respiratory tract infection (12.5% each).33 Persistent acrocyanosis/cyanosis and infusion-related reactions occurred in 8.3% of patients.33 In the CADENZA trial, one older patient with a history of diabetes had a severe cerebral venous thrombosis on day 86; this was assessed by the investigator as possibly related to sutimlimab.7 In the CARDINAL trial, no patients had thromboembolism during the study.6 In this study, dimers and thrombin–antithrombin were both reduced from baseline to week 26 (mean 846 ng/mL to 335 ng/mL and 16.6 μg/mL to 11.0 μg/mL, respectively) with sutimlimab treatment.6 Serious infections with encapsulated bacteria (Streptococcus pyogenes, Staphylococcus aureus and Staphylococcus epidermidis) were reported in the CADENZA trial, but no meningococcal infections or TEAEs of meningitis were identified in either study.6,7 Although serious infections were reported, there were no study discontinuations due to an infection.6,7
In summary, sutimlimab is a new and potent inhibitor of the classical complement pathway. Data from the pivotal clinical trials in CAD demonstrate excellent responses, with a significant reduction in anaemia, transfusion requirements, haemolysis, markers of inflammation and haemostatic activation.6,7 Sutimlimab was well tolerated with a good safety profile, and no Neisseria meningitidis infections were reported in either pivotal trial.6,7 Recent data suggest complement may contribute to refractoriness in other antibody-mediated disorders such as immune thrombocytopenic purpura. Sutimlimab may have a role in these disorders.34