New cancer treatments continue to move away from traditional cytotoxic chemotherapy to more specific and targeted therapies. One of the many striking examples of successful targeted therapy came from the identification of the bcr-abl fusion protein, the driving oncogenic force in chronic myeloid leukaemia (CML).1 Imatinib mesylate (Gleevec) was designed as a competitive inhibitor of the bcr-abl fusion protein,thus reversing the malignant CML phenotype. Although most other cancer types have more than one pathway involved, research is now focused on identifying critical up- or downregulated pathways andselectively targeting key molecules responsible for perpetuating the oncogenic state. One such promising target is survivin, a small 16.5kDa protein belonging to the inhibitor-of-apoptosis (IAP) family.2 Interest in survivin stems from its pattern of expression: it is upregulated in almost all tumour types while being minimally expressed in terminally differentiated tissues.3 Moreover, it plays a key role in both apoptosis and control of cell-cycle progression, and in tumours its expression is associated with poorer prognosis and increased treatment resistance.4 A number of strategies to inhibit survivin expression are being evaluated in the pre-clinical setting and some survivin-targeted therapies have now reached first-in-human trials. This article aims to review the role of survivin at a cellular and molecular level in normal and malignant tissues, the rationale for targeting the protein and the pre-clinical and early clinical data on survivin inhibitors.
Structure and Function of Survivin
The smallest member of the IAP family, survivin is composed of a single baculovirus IAP repeat (BIR) domain and an extended α-helical coiledcoil domain at the carboxy terminus5 (see Figure 1). It functions as a homodimer, requiring the BIR domain for dimerisation and recruitment of other proteins such as caspase 3, p21 and Cdk4.6 Differential splicing of the survivin gene pre-messenger RNA (mRNA) yields five distinct proteins: survivin (wild-type), survivin-ΔEx3, survivin-2β,7 survivin- 3β8 and survivin-2α.9 While wild-type survivin is often the predominant transcript, these survivin variants have been shown to have distinct functions and be of prognostic significance,10 e.g. delta Exon 3 and survivin-3β are cytoprotective,11 while survivin-2α and survivin-2β are pro-apoptotic.7 Delta Exon 3 has been associated with a poorer prognosis, higher stage or more aggressive disease in several tumour types,12,13 while low levels of survivin-2β correlate with high-stage gastric cancer10 and poorer prognosis in non-small-cell lung cancer (NSCLC).13
Role of Survivin in Apoptosis
Many studies have shown that survivin opposes both intrinsic and extrinsic mediators of apoptosis.4 Like most other IAPs, it acts upstream of effector caspases rather than through direct caspase inhibition.14 In the cytosol, survivin associates with hepatitis B X-interacting protein (HBXIP) and the complex binds to pro-caspase 9, preventing its recruitment and activation by the apoptosome.15 Another mechanism involves the sequestration of second mitochondria-derived activator of caspases/direct inhibitor of apoptosis-binding protein with low pI (Smac/DIABLO). Smac/DIABLO is a mitochondrial pro-apoptotic signal that binds X-linked IAP (XIAP), thereby preventing apoptosis inhibition. Survivin has been shown to prevent the release of Smac/DIABLO from the mitochondria, leaving XIAP free to inhibit caspase activity.16 Lastly, survivin can also bind directly to XIAP, stabilising it against degradation and resulting in inhibition of apoptosis.17
Role of Survivin in the Cell Cycle
Survivin is involved with all stages of the cell cycle from spindle formation to chromosome separation and cytokinesis. It functions, in part, as a member of the chromosomal passenger complex (CPC).18 In addition to survivin, the CPC comprises Aurora kinase B (AURKB) and borealin and inner centromere protein (INCENP), and has a major role in several aspects of cell-cycle control including spindle formation, kinetochore-microtubule attachments, the spindle checkpoint and cytokinesis.18,19 Survivin targets the CPC to various locations during cell division by means of its different domains. Survivin’s BIR domain localises the CPC to the centromeres, whereas its C-terminus domain localises it to the central spindle and mid-body.19 At the spindle checkpoint, survivin plays a key role as part of the CPC in retaining the anaphase-promoting complex/cyclosome (APC/C) inhibitor BubR1 at the kinetochores until correct microtubule attachments have been achieved.20 Survivin also operates independently of the CPC. It binds directly to microtubules in metaphase and anaphase, and is involved in regulation of microtubule dynamics.21
Regulation of Survivin
The survivin gene is regulated by a number of transcription factors including E2F, Sp1, β catenin, activated T-cell factor (TCF), signal transducer and activator of transcription (Stat)-3, hypoxia-inducible factor-1 alpha (HIF-1α), and heat shock protein (Hsp) 90.22 Factors upstream also play an important regulatory role. One study found that in normal melanocytes, p53 and retinoblastoma (Rb) are required to repress survivin transcription, with Rb exerting its effects through E2F and p53 repressing Sp1-mediated expression.23 Post-translational modifications also play an important regulatory role. Phosphorylation of Thr34 by p34cdc2-cyclin B1 slows down survivin clearance by the proteasome.22 Phosphorylation of Thr117 (by AURKB) regulates survivin’s cell-cycle activities, while phosphorylation of Ser20 (by protein kinase A [PKA]) facilitates the interaction of survivin with XIAP.24,25 Several sites of ubiquitination have also been identified and these include Lys23, Lys62, Lys78 and Lys79.26 Finally, Hsp90 acts as an important chaperone for survivin, necessary to prevent proteasomal degradation.27
Survivin and Cancer
Survivin is almost undetectable in most adult tissues, and expression is largely limited to developing embryos and haematopoietic, epithelial and gonadal cell lines, where expression is often cell-cycle-dependent.3 However, survivin over-expression has been reported in nearly all human malignancies.14 In most cases, this results from non-cell-cycledependent mechanisms driving survivin gene transcription. Three key intracellular pathways converge on the survivin promoter: the phosphatidylinositide 3,4,5-triphosphate kinase (PI3k)/Akt pathway,28 the Janus kinase (JAK)/Stat-3 pathway29 and the TCF/β-catenin pathway.30 The upregulation of these pathways is either in response to growth factors such as epidermal growth factor (EGF),31 interleukin (IL)- 6 or granulocyte-macrophage colony-stimulating factor (GM-CSF),32 or through downregulation of tumour suppressors such as adenomatous polyposis coli (APC),33 p53,34 promyelocytic leukaemia protein (PML)-435 and fragile histidine triad gene (FHIT).36 In fact, all of the best characterised tumour suppressor networks, such as p53, target the survivin gene, thus underscoring survivin’s importance in malignancy.37
Methods of Targeting Survivin and
Pre-clinical Results
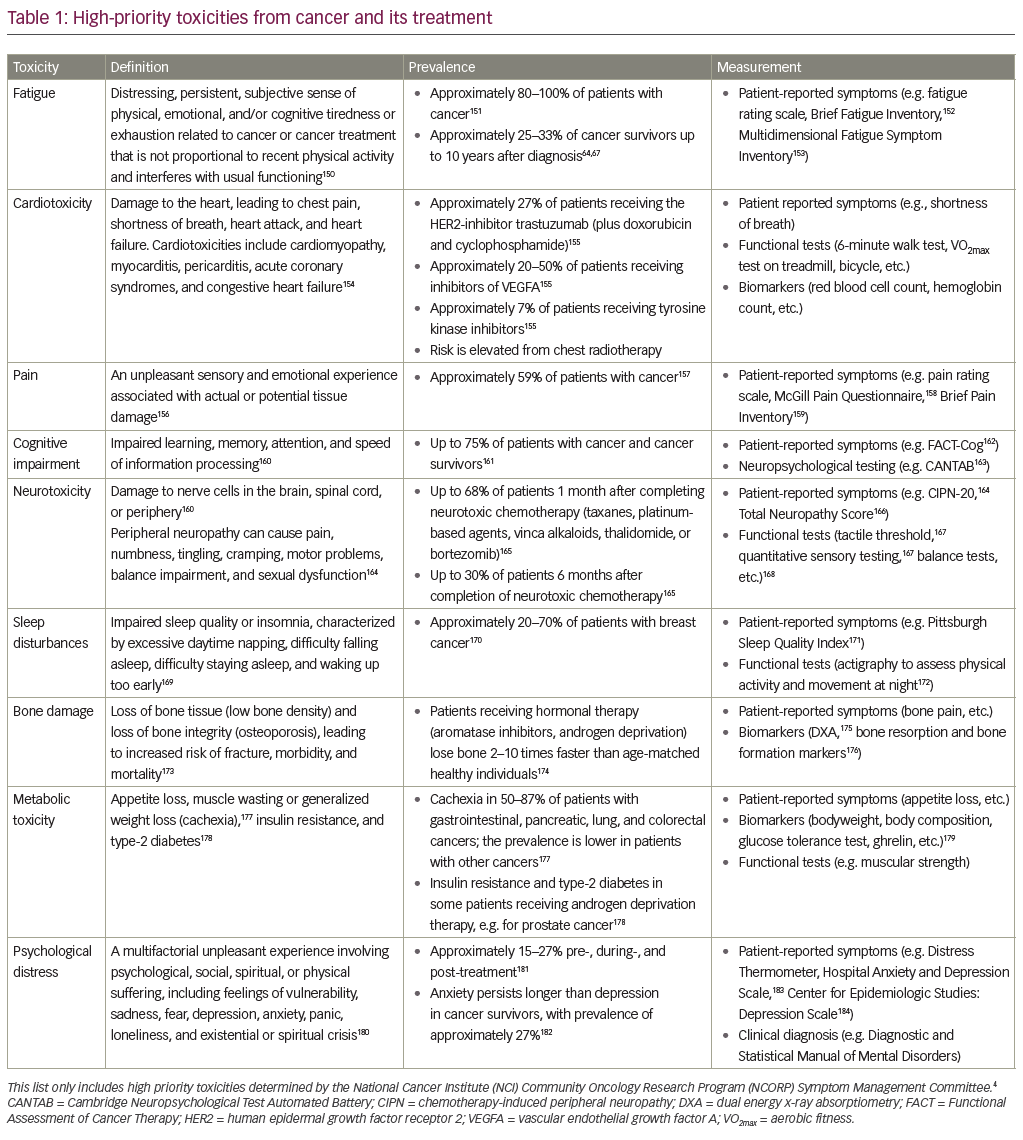
By knocking out survivin, multiple networks of cell proliferation and cytoprotection can be simultaneously disrupted. Direct survivin inhibition has been investigated at several levels: gene transcription, translation and protein degradation. Additionally, gene therapies and immunotherapies are in development (see Figure 2 and Table 1).
Transcriptional Inhibition
Small molecule transcriptional repressors have been developed. These include YM-155 and EM-1421 (tetra-O-methyl nordihydro-guaiaretic acid, terameprocol). YM-155 is an imidazolium-based molecule that inhibits survivin gene transcription through promoter binding, with consequent pro-apoptotic and antitumour effects on prostate cancer models.38 Phase I trials have demonstrated that YM-155 is well tolerated,39,40 and a phase II trial in 37 patients with previously treated advanced NSCLC reported two (5%, 95% confidence interval [CI] 1–18%) partial responses (PRs) and 14 (38%) patients achieving stable disease (SD), resulting in a disease control rate of 43% (95% CI 27–61%). Median duration of progression-free survival (PFS) was 1.7 months (95% CI 1.3 to 2.8 months), and median duration of overall survival (OS) was 6.6 months (95% CI 4–12.2 months). Treatment was well-tolerated.41 However, another phase II study in 34 chemotherapynaïve patients with unresectable stage III or IV melanoma reported only one PR, thus failing to meet its pre-specified primary end-point for efficacy of two responders in 29 evaluable patients.42 EM-1421 is a small-molecule transcription inhibitor that selectively targets the Sp1-dependent promoters of various genes, including CDC2 and survivin.43 This molecule has reached phase I trials as a vaginal ointment for use in human papillomavirus (HPV)-linked cervical intraepithelial neoplasia, and has an excellent safety profile.44,45 Moreover, a systemic formula of EM-1421 has also been developed, with promising antitumour effects in mouse models.43
Translational Inhibition
Molecular antagonists including antisense oligonucleotides (ASOs), hammerhead ribozymes and small interfering mRNA (siRNA) have been developed to target survivin at the level of mRNA translation. Grossman et al.46 were the first to use a survivin ASO against malignant melanoma cell lines. Transfection with the ASO triggered spontaneous apoptosis, and two-colour flow cytometry confirmed decreased endogenous survivin expression. Other pre-clinical studies confirmed these results47,48 and found that survivin downregulation sensitises certain human cancer cell lines to cisplatin and etoposide,49 as well as to radiotherapy.50
LY-2181308 is a second-generation ASO. Pre-clinical studies have demonstrated that LY-2181308 induces apoptosis and significantly reduces growth of xenografts.50 In the first-in-human phase I study, Talbot et al.51,52 reported that the ASO accumulated preferentially in tumour tissue, as shown by immunohistochemistry (IHC) and [11C]LY- 2181308 PET imaging. Reduced survivin gene expression and a 20–50% reduction in survivin protein levels were seen in 11 of 15 evaluable patients treated with LY-2181308, including near-complete elimination of survivin-positive cells in endobronchial tumour samples in two of three NSCLC patients. Restoration of apoptosis was demonstrated by elevation of cleaved caspase C3 and reduced Ki67 shown by IHC. LY2181308 was well-tolerated, with the most frequent adverse events being a transient prolongation of activated partial thromboplastin time (not associated with bleeding) and flu-like symptoms. A recent advance in ASO technology has been the development of locked nucleic acids (LNAs). LNAs contain a methylene bridge connecting the 2’-oxygen of ribose with the 4’-carbon. This modification increases serum stability and binding to complementary mRNA compared with earlier generations of ASOs. SPC3042 is the first ASO with LNAs directed against survivin. Early in vitro experiments and in vivo xenograft models of prostate cancer have shown pronounced cellular apoptosis, increased sensitivity to taxol when treated with SPC3042 and downregulation of Bcl-2.53 Hammerhead ribozymes are another method of targeting survivin mRNA. In 2003 Choi et al.54 designed two hammerhead ribozymes (RZ-1 amd RZ-2) that cleaved survivin mRNA at positions 279 and 289. The ribozyme sequences were cloned into an adenovirus vector, and infection of the MCF-7 breast cancer line resulted in increased apoptosis and sensitivity to etoposide. In melanoma cell lines, ribozymes targeting the 3’ end of the GUC294 triplet of survivin mRNA significantly reduced survivin levels and increased sensitivity to the topoisomerase-I inhibitor topotecan.55 The therapeutic potential of ribozymes has been hindered by the lack of efficient delivery systems, but solutions are emerging such as attaching the ribozyme to the pRNA of the phi29 bacteriophage.56 siRNAs against survivin were first used by Carvalho et al.20 in cultured HeLa cells. No detectable survivin was found 60 hours after cultures were transfected with survivin-specific siRNA, and the survivin-depleted cells were delayed in mitosis. In melanoma cell lines, siRNA against survivin mRNA sensitised cells to Apo2L/TNF-related apoptosis-inducing ligand (TRAIL)- induced apoptosis.57 In human sarcoma cells, however, survivin specific siRNA increased cell-cycle arrest but had little effect on apoptosis,58 while in an endothelial cell model survivin-specific siRNA caused both increased apoptosis and cell-cycle arrest, resulting in growth inhibition and reduced capillary formation.59
Targeting the Survivin Protein
Various strategies have been developed to impair survivin protein function. These include introduction of a dominant-negative survivin mutant and disruption of survivin–protein interactions. The delivery of dominant-negative survivin mutants via a plasmid or viral vector to tumour cells inhibits the effects of wild-type survivin in a dosedependent manner. One of these mutants is survivin T34A, in which the threonine at position 34 is replaced by alanine, thus rendering it non-phosphorylatable by p34cdc2-cyclin B1 and, consequently, preventing it from inhibiting apoptosis.60 The introduction of this mutant was shown to trigger spontaneous apoptosis in a number of cancer cell lines, including breast, cervical, prostate, colorectal and melanoma, as well as reducing tumour formation and growth in mouse melanoma and breast cancer models.61,62 Another mutant is survivin C84A, in which the cysteine residue is exchanged for alanine in the BIR domain. Survivin C84A displaces wild-type survivin from polymerised microtubules and completely abolishes survivin’s antiapoptotic activity.63 Introduction of this mutant into melanoma and colorectal cell lines caused an increase in apoptosis,46,64 and follow-up in vivo experiments demonstrated that survivin C84A reduced both tumour growth and angiogenesis, as well as enhancing the effects of 5-fluorouracil.64 Due to the survivin protein’s need for Hsp90 for stability, the peptidomimetic Shepherdin was developed to disrupt this interaction by interacting with the ATP-binding pocket on Hsp90. Early in vitro experiments showed that Shepherdin induces cell death within tumour cells, while sparing normal cells.65 In both breast cancer and acute myeloid leukaemia (AML) mouse xenograft models, Shepherdin was well-tolerated and inhibited tumour growth.65,66 After it was shown that Shepherdin residues 79–83 were essential for Hsp90 binding, a five-residue peptide called Shepherdin[79–83] with this same sequence was synthesised. Studies in AML cell lines showed rapid and complete killing of AML cell types and myeloblasts, but no effect of Shepherdin[79–83] on normal mononuclear cells.65 The non-peptidic small molecule 5-aminoimidazole-4-carboxamide-1- beta-D-ribofuranoside (AICAR) is another inhibitor of Hsp90. It mimics Shepherdin, binding the Hsp90 N-terminal domain. It has been shown to exert antiproliferative and proapoptotic activity in multiple tumour cell lines while having no effect on normal human fibroblasts.67
Cyclin-dependent kinase (CDK) inhibitors mediate their antitumour effects in part through their actions on survivin. Inhibition of p34cdc2- cyclin B1 prevents phosphorylation of survivin (Thr34), thereby reducing protein stability. In human hepatocellular carcinoma cells, flavopiridol was shown to increase TRAIL-induced apoptosis by upregulation of TRAIL receptors and downregulation of survivin.68 Purvalanol A, another CDK inhibitor, was also shown to induce apoptosis by inhibiting the JAK/Stat-3 pathway in MKN45 cells.69 A novel CDK inhibitor, NU6140, was shown to induce cell-cycle arrest and increase apoptosis in a concentration dependent manner. Moreover, a significant synergistic effect was observed when combining NU6140 and paclitaxel in HeLa cervical carcinoma cells.70
Targeting the Survivin Promoter
The high level of survivin expression in tumour cells has led to the idea of using the survivin promoter to drive expression of therapeutic genes, an approach still in early pre-clinical development. In 2005 Kamizono et al.71 reported having constructed a survivin-responsive conditionally replicating adenovirus (Surv.CRA), in which expression of the early region 1A (E1A) gene was regulated by the surviving promoter. Another group modified the mifepristone/RU486-regulated system to express a dominant-negative mutant of survivin (surDN) in colorectal tumour cells.72 Sher et al.73 constructed a plasmid containing a Bcl2 interacting killer (Bik) mutant gene, BikDD, whose expression was driven by the survivin promoter and resulted in selective killing of lung cancer cells both in vitro and in vivo.
Immunotherapy
A number of research groups have tested survivin-directed immunological approaches in pre-clinical models. For example, a survivin DNA vaccine was evaluated in murine pancreatic and lymphoma models, and was found to significantly slow tumour growth and prolong survival through increased lymphocyte infiltration at the tumour sites.74 In an NSCLC mouse model, oral delivery of a DNA vaccine triggered activation of antigen-presenting dendritic cells (DCs) and a cytotoxic T-lymphocyte (CTL) response, which resulted in elimination or suppression of pulmonary metastases.75 Another group developed a novel mimovirus vaccine, consisting of a cationic peptide containing a survivin CTL epitope and a plasmid encoding the murine IL-15 gene. The mimovirus was shown to inhibit tumour growth and prolong life in a mouse model.76 Survivin vaccines are now moving into the clinic. Peptide vaccinations have been used in colorectal cancers,77 urothelial cancers78 and breast cancers,79 all of which have been well tolerated. In one case report, a patient with refractory pancreatic cancer achieved complete remission for eight months after receiving peptide vaccinations consisting of a modified HLA-A2 restricted survivin (96–104) epitope.80
Another technique involves inducing survivin-specific CTLs with DCs. One group transduced DCs with the survivin gene via an adenoviral vector, and used these to induce CTLs with cytotoxic activity in urological cancer cell lines.81 In a phase I trial, a vaccination consisting of DCs loaded with five different HLA-A*0201-restricted peptides derived from prostate-cancer-associated antigens including survivin was evaluated. Of the eight patients with hormone-refractory prostate cancer treated, there was one PR and three patients had stable prostate-specific antigen (PSA) values or decelerated PSA increases. No side effects were noted other than local skin reactions.82
Conclusions
Because of its critical mitotic and anti-apoptotic roles, as well as its high levels of expression in many types of cancer, survivin is an excellent therapeutic target. A wide variety of strategies have been employed to reduce survivin activity, and strong pre-clinical data have led to early-phase clinical trials, which have thus far not reported any significant toxicities. The exact extent of the antitumour effect of these different strategies will become clear as data from phase II and III trials become available. If efficacy is demonstrated, survivin-directed therapies could be incorporated into a broad spectrum of oncological practice either as single agents or, more likely, in combination with existing chemotherapeutics, particularly those that exert their effects through apoptosis. Additionally, tumour survivin expression and change following therapy have the potential for use as predictive biomarkers to identify those patients most likely to benefit. ■