
To date, the cornerstone of curative treatment in early-stage gastrointestinal (GI) cancers includes surgery and sometimes radiation therapy. Depending on the stage and tumour type, these approaches are often combined with systemic neoadjuvant or adjuvant therapies. After all definitive therapy has been administered, physicians and patients rely on imaging and non-specific blood tests to monitor for recurrent disease while hoping for a cure. Unfortunately, many patients relapse and have more advanced, non-curable disease by the time recurrent cancer is measurable using imaging. One proposed strategy to potentially improve survival outcomes is to identify and eradicate cancer at its earliest sign of relapse. Liquid biopsy, or blood-based testing to identify circulating tumour components, is an increasingly studied tool for detecting minimal residual disease (MRD) – that is, persistent cancer at the microscopic level that is not yet large enough to be visualized directly or radiographically. It would be useful to be able to confirm both the presence or absence of MRD; the former could guide the augmentation of therapy, while the latter could help identify cured patients, reassuring them and sparing them toxicity from unnecessary further therapy (Figure 1).
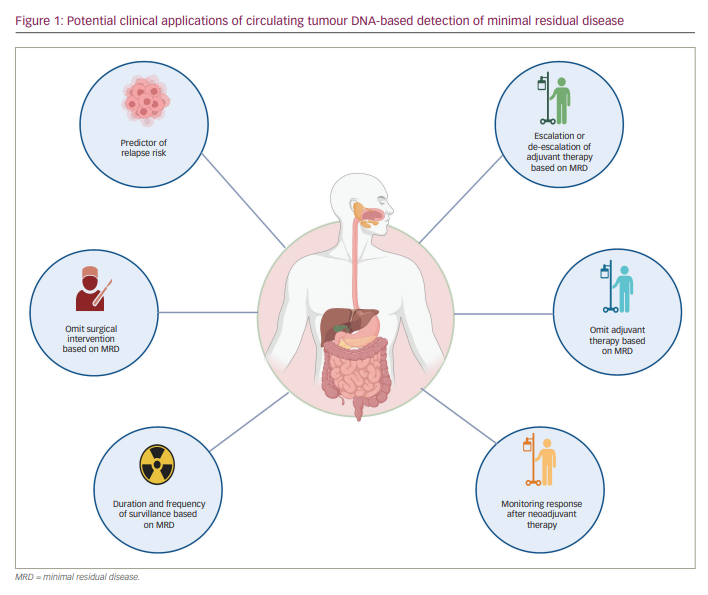
For example, the decision to offer adjuvant chemotherapy for patients with resected stage II/III colorectal cancer (CRC) is complicated, requiring a careful balancing of the risk and benefit for the individual patient. Adjuvant chemotherapy is associated with an overall survival (OS) benefit in patients with stage III CRC and some patients with stage II CRC, but it is difficult to predict the benefit for an individual patient.1,2 Features associated with high-risk stage II disease include poorly differentiated tumours, the presence of lymphovascular or perineural invasion, obstruction, perforation, positive margins, tumour budding and fewer than 12 lymph nodes examined.3 However, these criteria are more prognostic than predictive of benefit from adjuvant chemotherapy. Recommendations for adjuvant therapy in stage II disease vary, ranging all the way from observation only to 6 months of fluorouracil, leucovorin and oxaliplatin.3 As medical oncologists, we are generally biased towards over- rather than undertreating patients so as not to miss the opportunity to deliver potentially curative therapy. Clearly, we need to do better in terms of risk-stratifying patients with early-stage CRC and predicting who needs chemotherapy, how much and for how long. The same need for improved prognostic and predictive biomarkers can be seen across other GI tumour types and in other clinical settings, such as risk-stratifying patients with upper GI and rectal cancers managed with non-surgical wait-and-watch strategies. Until now, we have relied on what is essentially 19th-century technology (the pathology report) to guide therapy decisions, when what we really need is a reliable test for MRD. Circulating tumour DNA (ctDNA) is a promising tool to meet this need.
Cell-free DNA (cfDNA) refers to acellular fragments of double-stranded DNA released from cells that circulate in body fluids, including peripheral blood. While cfDNA was initially described in the 1940s, it was only after technological advances in the 1990s that we saw an exponential growth in its study.4,5 These fragmented DNA strands vary in length from 80 to 200 base pairs and can be released during cell death through apoptosis and necrosis, or active transport and spontaneous release from intact cells.6,7 The fragments circulate in the blood as free DNA, protein-bound DNA or DNA encapsulated by extracellular vesicles; they are present in higher levels in patients with conditions associated with more cell death, such as inflammation, trauma, autoimmune diseases and malignancy.5,8
ctDNA refers to cfDNA specifically released from tumour cells and is typically shorter than normal cfDNA. ctDNA accounts for a wide range – <0.1% to >90% – of cfDNA and has a half-life of 16 minutes to 2 hours.9–12 It is identified by detecting tumour-specific mutations or epigenetic changes in the blood through DNA sequencing and profiling. This DNA can be quantified, analysed for single or broad gene profiling, and utilized for real-time assessments of tumour dynamics based on its short half-life. It is increasingly being studied as a powerful, non-invasive tool for detecting early cancer and providing a prognosis. It might also reveal resistance mutations and provide an understanding of patient response to treatment.
ctDNA is emerging as a powerful tool in detecting MRD, the earliest sign of residual or recurrent cancer in low-volume, microscopic states, after curative-intent treatments such as surgery and adjuvant therapy. The concept of identifying MRD to prognosticate patients and guide standard-of-care treatments has already been established in haematological malignancies.13–17 For example, malignant haematologists use quantitative polymerase chain reaction (qPCR) to detect persistent chronic myeloid leukaemia using BCR-ABL1 transcript levels.18 Unfortunately, solid tumours manifest a diverse array of mutational events with significant inter- and intra-tumour heterogeneity that is not amenable to simple qPCR testing of one transcript. Ideally, an MRD test would be so sensitive and specific as to predict who can safely be observed without adjuvant chemotherapy and who needs chemotherapy due to a predicted survival benefit. With advancing genomic technologies, we are finally seeing MRD testing make its way into the solid tumour space.
While variable across solid tumour studies, ctDNA detection after definitive therapy often mirrors the expected recurrence rates, correlates with other prognostic clinicopathological features, and strongly prognosticates for relapse and survival while outperforming standard clinicopathological risk factors in observational studies.19–23 Emerging data also suggest that ctDNA clearance with adjuvant therapy is associated with improved survival, possibly representing micro-metastatic disease eradication. Still, prospective, randomized trial data are needed to validate the predictive value of this biomarker to guide adjuvant therapies.21,24
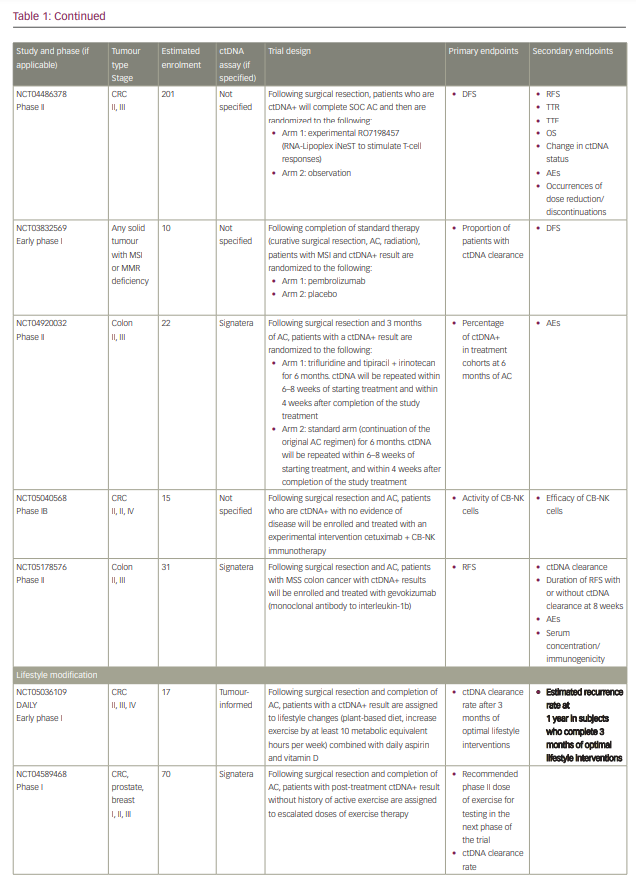
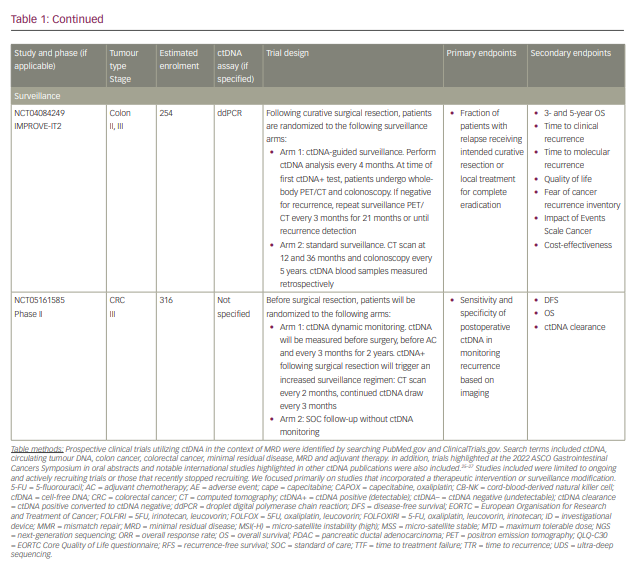
Commercialized ctDNA MRD assays are becoming more commonly used to provide patient prognoses in clinical practice. These assays are also being integrated into clinical trials to evaluate their role in adjuvant therapy guidance (Table 1).25–27 Using a ctDNA approach in the neoadjuvant and adjuvant settings has the potential to risk-stratify patients and identify high-risk patients who may truly benefit from adjuvant treatment or intensification, and those who are at low risk or unlikely to benefit and therefore should be spared treatment toxicities. ctDNA has the potential to provide early readouts of therapeutic efficacy or inefficacy on a microscopic level, supported by data correlating ctDNA kinetics with radiographic responses and survival outcomes.20,21,24,28–30
It has also essentially identified a ‘new clinical stage’ of disease post-treatment; one that involves no radiographic evidence of disease, but with detectable MRD. This new disease stage currently lacks any treatment guidelines. This previously undefined patient population identifies a high-risk group that may benefit from the development of novel treatment strategies to improve cure rates and cancer outcomes. However, most of the data today are derived from observational cohort studies, and many questions remain unanswered regarding the interpretation of different test results, strengths and limitations of various assays, the ability of ctDNA to guide adjuvant or post-adjuvant therapy, the clinical significance of rise-and-fall rates or stability of ctDNA, and the ability to individualize follow-up treatment. While we may not be there yet, in this review we describe the current state-of-the-art of blood-based MRD testing using ctDNA in CRC and other GI malignancies.
Circulating tumour DNA detection methods
While tumour tissue yields micrograms of DNA for genomic analysis, blood-based biopsies yield significantly lower amounts: reportedly in the range of nanograms cfDNA per 10 mL blood.31 A major challenge in blood-based testing for MRD relates to assay limits of detection; mean mutant allele frequencies (MAFs) or variant allele frequencies (VAFs) may be present at very low levels in relation to background cfDNA (<0.01%). One major approach to ctDNA detection is to use targeted assays to identify single or limited pre-specified tumour mutations. A second approach is to use broader, non-targeted, genome-wide sequencing to capture multiple genes.
For targeted approaches, digital PCR (dPCR) techniques have emerged as more robust and precise methods for detecting low rates of mutations compared with qPCR. dPCR includes digital droplet PCR (ddPCR) and beads, emulsion, amplification and magnets (BEAMing) techniques, both of which can identify pre-specified, known mutations with high accuracy in the plasma. ddPCR uses a water–oil emulsion technology and distributes DNA fragments into thousands of droplets, each of which contains a single DNA strand, and uses PCR and validated probes to detect and quantify mutations. It is a highly sensitive test for low concentrations of nucleic acids, but requires defining pre-specified mutations for interrogation and can only evaluate a limited number of genes at a time.32,33 This method has successfully detected MRD and provided GI cancer patient prognoses.34–36 While more expensive and involved, the BEAMing method combines dPCR with magnetic beads and flow cytometry to deliver an even more sensitive test for detecting and quantifying a small number of targeted mutations that may be present at very low frequencies (0.01%).37
In contrast, broader applications, such as next-generation sequencing (NGS), can more completely analyse cfDNA and uncover multiple mutations across a broad panel of genes. However, unlike targeted PCR-based assays, which can detect VAFs with deep sequencing in the range of 0.001–0.01%, non-targeted NGS assays achieve broad interrogation at the cost of lower sensitivity and detect VAFs in the range of 0.02–10%.31,32,38–40 Multiple NGS platforms exist, and their strengths and limitations have been well described in the literature.32,41 For example, the upgraded multiplex PCR-based NGS TAm-Seq technology, which utilizes tagged-amplicon deep sequencing to target multiple pre-specified genes on a panel, can detect single-nucleotide variants, short insertions and deletions, and copy number variants with high sensitivity at 0.25–0.33% allele frequencies, and can detect mutations at even lower levels at a 0.02% allele frequency.42,43 Although this upgraded technology can interrogate multiple genes and is relatively inexpensive, it also requires prior knowledge of targeted mutations.41 Massive parallel sequencing platforms are prone to error during the sequencing process. Other assays, such as Safe-SeqS, in which unique identifiers are added to template DNA molecules prior to amplification, incorporate error-reducing strategies to improve sensitivity.44 Another platform, CAPP-Seq, uses hybrid-capture to detect all major mutation classes and includes rearrangements that are not always detected on other amplicon-based platforms.45
Whole-exome sequencing (WES) and whole-genome sequencing allow for the most comprehensive profiling, interrogate the whole exome or genome, and do not limit sequencing to predetermined genes. However, this breadth of coverage comes at the cost of higher expense, lower sequencing depth and lower sensitivity.41 These differences are essential to consider when choosing an optimal assay for MRD detection, where robust sensitivity is critical to reducing false-negative results. Methods such as template tagging, duplex sequencing, single-molecule sequencing, error-correcting algorithms, incorporating epigenetic marker evaluation and enriching for highly fragmented ctDNA, among others, are being used to improve the ability to detect low-level MAFs with NGS.38,40,46
Minimal residual disease detection methods
While the techniques to detect ctDNA may vary, MRD assays can be broadly characterized as either tumour agnostic or tumour informed. Tumour-agnostic assays, also known as plasma-only assays, do not require a prior tissue biopsy or knowledge of a patient’s tumour genomic profile; they either narrowly or broadly interrogate cfDNA for commonly mutated cancer genes. Although tumour-agnostic assays may be less sensitive than tumour-informed assays due to their use of broad panels, which compromises the depth of sequencing, they are quick (7- to 10-day turnaround), relatively inexpensive and more comprehensive in characterizing mutations and identifying emerging resistance mechanisms on serial analysis. Tumour-agnostic assays have been applied in CRC MRD studies and have demonstrated prognostic value in predicting relapse and survival.19,35,46 For example, Parikh et al. recently reported a recurrence sensitivity and specificity of 55.6% and 100%, respectively, using the Guardant Reveal/LUNAR-1 plasma-only test in prospectively studied patients with CRC who underwent definitive surgery.46 In this study, the sensitivities improved with longitudinal testing and integration of epigenomic signatures in testing. Tumour-agnostic assays have also demonstrated prognostic value in other prospective and retrospective studies in resected upper GI and pancreatic cancers.23,47–49 When choosing MRD tests, it is important to realize that tumour-agnostic, NGS-based assays for MRD may differ from those frequently used to evaluate mutational profiles in metastatic disease. For instance, MRD assays may specifically combine DNA with epigenetic marker analyses or other methods to increase sensitivity in detecting minuscule ctDNA levels.46
In contrast, tumour-informed assays require prior knowledge of a tumour’s mutational profile through tissue analysis to design patient- and tumour-specific assays targeting these mutations in the blood. For example, the Signatera ctDNA test, which is commercialized and has received US Food and Drug Administration (FDA) Breakthrough Device Designation, requires WES of a patient’s primary tumour and the buffy coat to identify 16 clonal tumour-specific somatic mutations that are not driver mutations under selection pressure and that are not present in the germline.50,51 Sixteen PCR primers are designed for the identified single-nucleotide variants, and a patient’s peripheral blood is drawn at pre-specified times and then tested against the multiplex PCR with ultra-deep sequencing. If at least two mutations are detected, this results in a positive read. This assay can detect very low fractions of tumour DNA (<0.01%) with >95% sensitivity. While other tumour-informed assays using Safe-SeqS, ddPCR and other designs exist, the Signatera test has been widely studied in many GI and non-GI tumour types, demonstrating impressive sensitivities (88–100%) and specificities (93–100%) for MRD, positive predictive values for relapse (>98%) and median lead times from ctDNA detection to relapse (8–9 months).20–22,24,29,34,36,50,52–65 These highly specific and sensitive assays are particularly attractive for MRD detection and are being increasingly studied in this space. However, due to the inherent process of tumour, germline and then peripheral blood sequencing, tumour-informed testing may be more expensive than the plasma-only approach, as it first requires tumour tissue acquisition and testing, and takes longer to obtain results (4–6 weeks for the first and then 7–10 days for subsequent test results). In addition, if there is insufficient tumour tissue, these tests may not be feasible. For example, creating the commercialized Signatera test requires WES on tumour samples with a surface area >25 mm,2 volume ≥60 microns in thickness and tumour content ≥30% when submitted as formalin-fixed paraffin-embedded tumour blocks or unstained slides.66 Fine-needle aspirations, resected tumour with significant treatment effect and scant cytology samples may not always yield adequate tissue for these tests.
Considerations in circulating tumour DNA interpretation
Although the data today increasingly support ctDNA as a powerful tool in the MRD setting, we must remember it is not a perfect test. We have already described some of the inherent sensitivity and technical limitations of various assays. It is important to counsel patients on the potential for false negatives when there may, in fact, be residual disease that is below an assay’s detection threshold. Some studies have demonstrated that testing sensitivities improve with serial monitoring.29 Therefore, it is important to consider longitudinal testing in the adjuvant or post-adjuvant setting in case a negative test turns positive after the cancer has been given enough time to grow. In addition, assays that target one or a few genes may miss subclones of heterogeneous tumours. Testing multiple genes or selecting for early clonal mutations that are not under selection pressure, as seen in the Signatera assay, may overcome these limitations.67 The exact testing intervals and total duration for which we should follow patients with negative tests are currently unknown.
ctDNA detection and quantification can also be influenced by multiple factors such as the primary tumour type, staging, assays used, mutations analysed and organs involved. For example, Bettegowda et al. demonstrated that the frequency and concentration of detected ctDNA in metastatic cancers varied based on tumour type, with relatively higher frequencies of detection in bladder, colorectal, gastroesophageal and ovarian cancers; and lower frequencies of detection in central nervous system, renal, thyroid and prostate cancers.10 Although present at lower concentrations, ctDNA can also be detected in localized, early-stage cancers, and frequencies and concentrations trend higher with higher clinical stages.
Others have shown that the sites of metastatic disease and size of lesions may influence ctDNA levels, with higher detection and concentrations in patients with liver metastases compared with those with lung- and peritoneal-only metastases.68,69 For example, Van’t Erve et al. reported that ctDNA was detected in 93% of patients with CRC with liver metastases compared with 20% of those with peritoneal metastases.68 Bando et al. reported that ctDNA median VAFs were highest in patients with CRC liver metastases (23.1%), lower in those with lymph node metastases (6.0%) and lowest in those with lung and peritoneal metastases (0.4% in both).69 These findings suggest that different tumours have different potentials for ‘DNA shedding’ into the bloodstream, theoretically due to variations in tumour biology, tumour microenvironment and degree of vascularity, for example. Currently, we cannot use ctDNA to predict recurrence location and patterns, since ctDNA shedding may vary based on organ involvement. Therefore, it is possible that patients may develop recurrence in sites associated with ‘low shedding’ with false-negative ctDNA values. We also currently do not know how to interpret different rates of ctDNA changes or ctDNA stability, and how they translate to clinical management.
On the other hand, while many tests are highly specific, we must also be aware of possible false positives. For example, clonal haematopoiesis of indeterminate potential (CHIP) is a process that can be seen with normal aging and even in healthy individuals where there is clonal expansion of haematopoietic stem cells with accumulating somatic mutations in the absence of malignancy or haematological disorders.70 Some of these CHIP-related aberrations may involve genes often associated with GI malignancies, such as TP53 and RAS, among others, and may lead to false-positive results in MRD testing.71–73 To address this, some platforms use bioinformatics to filter out likely benign clonal variants, while others sequence matched tumour samples and peripheral blood leukocytes and perform ctDNA interrogation only for tumour-specific mutations that are not present in the buffy coat.57,74
With ctDNA tests such as Signatera, which has FDA Breakthrough Designation and Medicare coverage for patients with stage II/III CRC,75,76 and many other tests in the pipeline, clinicians are increasingly using ctDNA for MRD monitoring in routine practice. However, many clinicians, especially in the community, are understandably hesitant to incorporate this into routine management in the absence of prospective, randomized data to help guide treatment escalation or de-escalation. While these tests are demonstrating powerful prognostic value, questions remain regarding their predictive capabilities and what to do with the results. Several clinical trials using different assays have been developed to answer these questions, especially in CRC (Table 1). We need large patient numbers to validate survival endpoints when using ctDNA-guided strategies, and increasing community and academic trial enrolment and awareness will hopefully facilitate this research.
Minimal residual disease in colorectal cancer
For patients with stage II/III CRC following surgical resection, the presence of ctDNA is clearly a negative prognostic factor. In an analysis of the IDEA-France trial by Taieb et al., 1,017 patients with stage III CRC and ctDNA suitable for analysis collected prior to adjuvant chemotherapy were followed for disease-free survival (DFS) and OS. Overall, 14% of patients had positive postoperative ctDNA, and positive ctDNA was associated with inferior DFS (3-year DFS rate 66.4% versus 76.7%; hazard ratio [HR] 1.46, 95% confidence interval [CI] 1.08–1.97; p=0.015) and OS (5-year OS rate 81% versus 87%; HR 1.56, 95% CI 1.08–2.26; p=0.018).35 Similar findings have been reported for carcinoembryonic antigen (CEA). Sonoda et al. looked at postoperative CEA in 482 patients with stage II/III CRC, and elevated postoperative CEA correlated with significantly inferior DFS and OS.77 However, only 7.6% of patients with stage II disease and 10.6% of those with stage III disease had elevated postoperative CEA, implying that CEA may be less sensitive than ctDNA. Indeed, a study of 110 Japanese patients with metastatic CRC showed a concordance rate of only 75.5% between CEA and ctDNA.78 Moreover, many tumours do not make CEA at baseline preoperatively. The question is: how do we adjust our adjuvant treatment strategies for positive or negative ctDNA? Stated another way, can we rely on negative ctDNA to predict a lack of benefit from adjuvant chemotherapy and positive ctDNA to predict a benefit? We need a test to predict who needs adjuvant chemotherapy and who does not.
Even if ctDNA is not yet able to predict who needs chemotherapy, a growing body of data suggests it may aid in the decision-making process, and ctDNA is the focus of several ongoing, prospective clinical trials to better answer these questions. While positive postoperative ctDNA is negative prognostically, clearance of ctDNA post-adjuvant chemotherapy may highlight a cohort that truly benefits from adjuvant chemotherapy.21,24,57
The most relevant clinical use of ctDNA in the current paradigm is serial monitoring. Even with the advent of ctDNA, standard surveillance includes imaging, laboratory work and visits through 5 years, in addition to surveillance colonoscopies.3 If nothing else, persistently negative longitudinal ctDNA may be the best early predictor of cure: in a Danish study of 125 patients with stage I–III CRC by Reinert et al., only two of 60 (3.3%) persistently ctDNA-negative patients experienced disease recurrence.57 Thus, patient anxiety, especially around the timing of surveillance scans, can hopefully be lessened somewhat as their odds of recurrence with persistently negative ctDNA results drop to relatively very low levels.
Stage II CRC may be the best setting for using ctDNA to guide therapeutic selection. Patients with low-risk stage II disease are typically suitable for observation alone, but a minority of these patients will be ctDNA positive postoperatively and have a higher risk of recurrence, and will potentially benefit from adjuvant chemotherapy.3 This strategy is being actively studied in the COBRA trial (NRG-GI005; ClinicalTrials.gov NCT04068103).79 Conversely, the benefit of adjuvant chemotherapy in high-risk stage II disease is often overstated, and many patients are overtreated. Thus, ctDNA may be useful to de-escalate adjuvant treatment in this population, where the guidelines recommend a wide range of choices: from 6 months of doublet chemotherapy to observation alone.3 Again, this strategy needs to be evaluated in robust, prospective clinical trials. While we are still learning how to harness the power of this new technology, anecdotal reports of how ctDNA has played a vital role in clinical decision-making in patients with stage II disease have been published.80
Another clinical quandary is whether to offer adjuvant chemotherapy to patients after metastasectomy for stage IV oligometastatic CRC. In the Japanese JCOG0603 trial, 300 patients with resectable liver metastases were randomized 1:1 to 6 months of adjuvant fluorouracil, leucovorin and oxaliplatin or surgery alone.81 While DFS was longer with chemotherapy (5-year rate 49.8% versus 38.7%; HR 0.67, 95% CI 0.50–0.92; p=0.006), there was no OS benefit (5-year rate 83.1% versus 71.2%; HR 1.25, 95% CI 0.78–2.00; p=0.42), and patients who had already received prior adjuvant chemotherapy after primary tumour resection tended to do worse in DFS and OS with more chemotherapy in subgroup analyses. In this setting, negative ctDNA post-metastasectomy may provide some extra solace that chemotherapy may not benefit this group, especially for less fit patients. Again, a prospective study to validate this would be preferred to determine whether positive ctDNA post-metastasectomy correlates with benefit from chemotherapy.
The IDEA collaboration established 3 months of adjuvant doublet chemotherapy as an appropriate alternative to the traditional 6 months of adjuvant therapy for patients with stage III CRC.82 What can ctDNA add to this decision-making algorithm? In the Taieb et al. analysis mentioned above, patients who were ctDNA positive postoperatively and received 3 months of adjuvant chemotherapy had significantly shorter DFS compared with ctDNA-positive patients treated with 6 months of therapy and ctDNA-negative patients treated with 3 or 6 months of therapy (p=0.0021). These findings were especially notable among patients with high-risk disease (T4 and/or N2, p<0.001), and while this trend was noted in low-risk disease, it was not statistically significant (p=0.47).35 It is important to recognize that these findings were retrospective, and future studies should prospectively address whether escalation from 3 to 6 months of adjuvant therapy is beneficial in the ctDNA-positive population. In practice, we are justifiably nervous when someone is postoperatively ctDNA positive, and it is very reasonable to continue adjuvant therapy to 6 months if the patient remains ctDNA positive after 3 months of adjuvant therapy. What is less clear is whether someone who clears ctDNA after 3 months will benefit from an additional 3 months of chemotherapy.
Emerging data from Japan highlight the potential predictive abilities of ctDNA. The observational GALAXY study in CIRCULATE-Japan analysed Signatera ctDNA testing in 1,040 patients with resected stage I–IV CRC, and demonstrated that positive ctDNA 4 weeks after surgery was negatively prognostic for DFS (HR 13.3, 95% CI 8.0–22.2; p<0.001) and that ctDNA trends at 12 weeks post-surgery were also prognostic with a worse DFS reported in patients who remained ctDNA positive compared with those who converted from positive to negative (HR 15.8, 95% CI 5.7–44.2; p<0.001).24 Most interesting was that patients with high-risk stage II or any stage III CRC with negative 4-week postoperative ctDNA received no benefit from adjuvant chemotherapy (adjusted HR 1.3, 95% CI 0.5–3.6; p=0.63), implying we are likely overtreating patients with negative ctDNA. With the important caveats that the decision to give chemotherapy was not randomized based on ctDNA and that the median follow-up time of 11.4 months is relatively short, GALAXY adds more evidence that we will likely be using ctDNA to guide adjuvant therapy decision-making in the near future.
ctDNA may also have important roles in the management of locally advanced rectal cancer (LARC), where curative-intent therapy is multimodal with chemotherapy, radiation and surgery. For instance, although adjuvant chemotherapy after neoadjuvant chemoradiation and surgery is recommended, the survival benefits are controversial, and questions remain regarding which patients truly benefit and what chemotherapy regimens are best used.83 Improved biomarkers to risk-stratify patients would be helpful in this space. Tie et al. prospectively collected blood samples for tumour-informed ctDNA analysis at baseline, after chemoradiation and after surgery in 159 patients with LARC, and reported worse recurrence-free survival (RFS) in patients with ctDNA detected after chemoradiation (HR 6.6, p<0.001) and after surgery (HR 13.0, p<0.001), with a more prominent prognostic impact in patients not treated with adjuvant therapy.53 In addition, a recent systematic review of 21 publications highlighted the promising prognostic potential of ctDNA in LARC.84 Another systematic review that reported on 25 published studies (eight of which specifically evaluated prognosis) also summarized the prognostic value of ctDNA detection at baseline, during or post-neoadjuvant therapy and post-surgery.85 These evolving data indicate that using ctDNA as a prognostic biomarker may help to identify high-risk patients in need of adjuvant treatment.
Another potential use for ctDNA is in patients with rectal cancers who are considering a watch-and-wait approach following neoadjuvant therapy. For patients with low-lying rectal cancers, the prospect of a permanent ostomy is frequently a non-starter when considering surgical resection. Given that some patients may have a pathological complete response and may not have required surgery, ctDNA could help risk-stratify this population and potentially tilt the scales in favour of surgery for those who are ctDNA positive. Recent studies summarized in two systematic reviews have demonstrated conflicting results on the ability of ctDNA to predict tumour responses and pathological complete response after chemoradiation, although the trial designs and ctDNA detection methods utilized were highly variable across studies.84,85 Combining ctDNA data such as present mutations, baseline levels and dynamics on therapy, along with other diagnostics findings such as tumour regression grading on MRI, has the potential to improve our ability to predict a pathological complete response or residual disease.86 While these approaches are encouraging, a lot of work needs to be done to elucidate the prognostic and predictive roles of ctDNA, especially when considering organ-preserving strategies or treatment de-escalation in LARC.
As discussed above, ctDNA clearly has multiple shortcomings, and there is a lot that we do not know. These include the fact that a negative ctDNA test, especially at a single postoperative time point, does not rule out MRD as it could be below the level of detection. We also do not know whether ctDNA can be used to influence decisions regarding the use and duration of adjuvant chemotherapy. However, there is a fair amount of retrospective evidence, and prospective trials are ongoing. Finally, ctDNA may allow us to alter our surveillance strategy; for example, intensifying surveillance for those who are ctDNA positive and spreading out surveillance for those who are ctDNA negative. However, we do not have the evidence to make these changes yet.
Minimal residual disease in pancreatic ductal adenocarcinoma
While ctDNA is best described in CRC, it is also gaining traction in other GI tumour types. Utilizing this technology to improve outcomes is particularly attractive in pancreatic ductal adenocarcinoma (PDAC), an aggressive and lethal disease with dismal survival rates.87 Despite advances in oncology, therapeutic options for patients with PDAC continued to be limited, making surgical resection the only hope for a cure. Although surgical techniques have improved and different treatment methods (including chemotherapy and radiotherapy) have been incorporated, the prognosis is still poor, with 5-year OS rates of 39% and 13% in localized and locally advanced disease, respectively.88 Due to the high rates of recurrence, both systemically (>80%) and locally (>20%) following curative-intent surgery, there is a pressing need for a prognostic marker that measures MRD.89
In PDAC, multiple studies have proved the prognostic value of serum or plasma ctDNA to detect MRD.49 Several techniques have been used to analyse ctDNA, including whole-genome sequencing, targeted gene sequencing and ddPCR.90 WES has not been studied widely, given its high cost and low sensitivity. As previously discussed, in contrast to WES, targeted gene sequencing and ddPCR can identify mutant ctDNA with MAFs as low as <0.2% and <0.1%, respectively, making them more sensitive tools for detecting mutant ctDNA and better methods for monitoring MRD.91,92 As PDAC has a high prevalence of KRAS proto-oncogene mutations, molecular profiling of resectable PDAC ctDNA samples has been largely limited to targeted gene sequencing and ddPCR to detect KRAS mutations.93
A prospective, multicentre biomarker study enrolled 42 patients with early-stage, operable PDAC and matched blood and tissue samples.54 PCR-based Safe-SeqS assays were used to obtain pre- and postoperative samples for ctDNA analysis. After a median follow-up of 38.4 months, the study found that detection of ctDNA preoperatively was associated with poorer RFS (HR 4.1, 95% CI 1.8–9.0; p=0.002) and OS (HR 4.1, 95% CI 1.6–10.5; p=0.015). Furthermore, the presence of ctDNA after curative-intent resection was also associated with worse RFS (HR 5.4, 95% CI 1.9–15.2; p<0.0001) and OS (HR 4.0, 95% CI 1.2–13.6; p=0.003). Notably, 13/13 (100%) patients with detectable ctDNA postoperatively experienced disease recurrence, regardless of adjuvant treatment, compared with 10/22 (45%) patients with undetectable postoperative ctDNA.
Another study has looked at the role of peri-operative KRAS-mutant ctDNA in resectable PDAC by collecting plasma samples from 31 patients and analysing ctDNA using ddPCR.94 Six of the 31 patients who had surgery tested positive for postoperative ctDNA. The presence of ctDNA after surgery was linked with significantly poorer RFS (4.6 versus 17.6 months; p=0.03) and OS (19.3 versus 32.2 months; p=0.027).
Our group recently conducted a comprehensive review and meta-analysis to assess the prognostic significance of KRAS-mutated ctDNA in resectable PDAC.95 A total of 13 studies and 954 patients were included in the final analysis. Compared with negative ctDNA, positive ctDNA in the preoperative setting was associated with worse RFS (HR 2.067, 95% CI 1.346–3.174; p<0.001) and OS (HR 2.170, 95% CI, 1.451–3.245; p<0.001). In the postoperative setting, positive ctDNA was again associated with worse RFS (HR 2.986, 95% CI 1.897–4.699; p<0.001) and OS (HR 5.812, 95% CI 1.757–19.228; p=0.004) compared with negative ctDNA.
At the American Society of Clinical Oncology GI 2022 conference, a prospective cohort study was presented that examined the prognostic value of a personalized and tumour-informed ctDNA test (Signatera bespoke multiplex PCR NGS assay) in 93 patients with PDAC.96 Postoperative ctDNA positivity at any time was associated with poorer RFS compared with ctDNA negativity (HR 8.0, 95% CI 3.4–18.7; p=1.6×10).6 Elevated cancer antigen (CA) 19-9, on the other hand, was not linked with RFS (p=0.35).
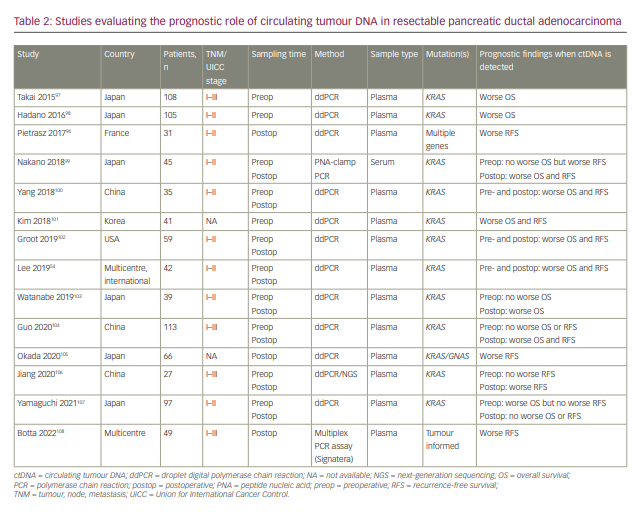
At the time of writing, there is growing evidence supporting the use of ctDNA as a marker of MRD in resectable PDAC (Table 2),54,94,97–108 which might have implications for the aggressiveness of monitoring and length of adjuvant therapy in PDAC. However, more prospective trials are needed to assess this utility.
Minimal residual disease in other gastrointestinal cancers
A limited number of studies have investigated whether ctDNA can be used as a predictive or prognostic tool in other cancers such as upper GI malignancies. One prospective study enrolled 97 patients with oesophageal adenocarcinoma treated with neoadjuvant chemotherapy and surgery to determine whether ctDNA was correlated with survival.65 Out of 77 patients with available postoperative ctDNA samples and a known cause of death, 36 (47%) had recurrent disease within 60 months of follow-up. Sixteen out of 77 patients (21%) were ctDNA positive following resection, and recurrence was observed in 12 of these patients (75%). Furthermore, the mean cancer-specific survival for ctDNA-positive patients was half that of ctDNA-negative patients (14.9 versus 29.5 months; HR 2.32, 95% CI 1.14–4.73; p=0.03), and ctDNA-positive patients also had worse DFS (HR 2.35, 95% CI 1.18–4.72; p=0.01). In another study of oesophageal cancers, Azad et al. tested for ctDNA before and after neoadjuvant chemoradiation, and reported that ctDNA detection after chemoradiation was associated with tumour progression, distant metastases and inferior survival.109 For patients who did not proceed with surgery, ctDNA combined with metabolic imaging analyses after chemoradiation was more sensitive than ctDNA alone or positron emission tomography–computed tomography alone in predicting recurrence (sensitivities of 100%, 71.4% and 57.1%, respectively).
In gastric cancer, one study has reported on 46 patients with stage I–III disease who underwent surgical resection with curative intent.23 The sensitivity and specificity of a positive, post-treatment ctDNA result predicting recurrence at 30 months were 39% and 100%, respectively. Additionally, ctDNA positivity after surgical resection was associated with statistically significantly worse DFS (HR 6.56, 95% CI 8.316–208.5; p<0.0001) and OS (HR 5.959, 95% CI 3.765–138.1; p=0.0007). Serial, longitudinal testing was also prognostic and identified emerging mutations during and after systemic therapy. Interestingly, this study also reported that ctDNA was detected a median of 179 days prior to radiographic evidence of recurrence. This opens the possibility for ctDNA to be used as a marker to intensify therapy earlier in these types of cancer. Recently, Huffman et al. used the Signatera tumour-informed MRD assay prospectively in 254 patients with oesophageal, gastro-oesophageal junction and gastric carcinomas. These researchers reported ctDNA positivity rates of 22.6–46.9% for resected localized disease and 62.5–100% for stage IV disease.110 MRD detection was similarly associated with worse survival outcomes. While these studies are beginning to demonstrate the prognostic potential of ctDNA in upper GI cancers, further data are needed to validate ctDNA as a tool to risk-stratify patients in controversial clinical scenarios in addition to detecting MRD after all definitive therapy. For example, ctDNA in combination with other diagnostics could potentially be applied to predict complete clinical or pathological responses after neoadjuvant therapies and select patients who may safely
be offered a wait-and-watch approach instead of proceeding with major oesophagogastrectomies.
ctDNA also has potential applications in hepatobiliary cancers, where we see inconsistent production of the non-specific protein tumour markers (CA 19-9 and alpha-fetoprotein) used currently to monitor disease. Kasi et al. were among the first to use a tumour-specific assay to study MRD in these cancers.111 Using Signatera, they reported 20–29% MRD rates across patients with hepatocellular and biliary carcinomas, with detection of MRD being significantly associated with disease stage. Additional data on longitudinal testing and associations with clinical response are pending. Another group had previously used a tumour-agnostic ddPCR method to detect hotspot mutations in patients with resected hepatocellular carcinoma, and also reported that increased postoperative ctDNA MAFs were associated with relapse and shorter survival.112
Smaller trials have broadened the application of ctDNA to other, less common types of GI cancers. In GI stromal tumours, early data suggest that ctDNA detection intraoperatively may correlate with tumour risk, ctDNA kinetics during systemic therapy and surgery may assess response, and ctDNA analysis may identify emerging tyrosine kinase inhibitor resistance mechanisms.113,114 However, robust studies evaluating the role of ctDNA in MRD in GI stromal tumours are currently lacking. In anal cancer, where concomitant human papillomavirus infection is highly prevalent, human papillomavirus ctDNA was recently studied as a biomarker, and detection after definitive chemoradiation was strongly associated with inferior DFS.115 This highlights the possibility of using ctDNA to measure MRD in other types of viral-induced cancers. Further research is needed to understand the applicability and feasibility of measuring residual disease in a wide variety of cancers.
Circulating tumour cells to assess minimal residual disease
In addition to ctDNA and cancer-derived protein markers, other liquid-biopsy components such as circulating tumour cells (CTCs) have the potential to detect residual disease. CTCs are tumour cells that have been shed from a primary or metastatic tumour into the bloodstream, and it is believed that subsets of these released from a primary tumour are responsible for metastases.116 Studies have demonstrated that the presence of CTCs is prognostic in advanced CRC and correlates with stage of disease.117–120 Studies in advanced CRC have also demonstrated decent concordance of PCR-based sequencing of CTCs, tumour tissue and ctDNA in hotspot mutations.121 Therefore, isolating and studying CTCs from peripheral blood may be another non-invasive way of detecting residual cancer after curative therapies and studying a tumour’s heterogeneous mutational landscape. In non-metastatic CRC, the presence of CTCs has been reported as a negative prognostic factor for progression-free survival and OS across many studies.119,122–124 In the adjuvant setting in CRC, the presence of postoperative CTCs has been reported as a poor prognostic factor for DFS and OS in some studies125,126 but as being non-prognostic in others.127
However, a notable limitation in utilizing CTCs today stems from the major challenge of isolating intact, rare CTCs from peripheral blood. CTC isolation techniques largely rely on antibodies for tumour markers and cell properties (e.g. size and density).128,129 For example, the FDA-approved CellSearch technology harvests epithelial cell adhesion molecule-positive cells, while also utilizing a leukocyte common antigen (CD45)-negative selection process to remove leukocytes.130 CellSearch technology and other methods that isolate cells based on physical properties have success rates of ≤25% in harvesting CTCs, although new strategies improving upon this are under way.129 In addition, genomic discordance has been reported among CTCs, tumour and ctDNA, and may reflect either heterogeneous tumour subclones or limited sensitivities of CTC genomic testing in samples with low CTC yield and purity.121
Fortunately, in the past decade, we have seen a rise in CTC studies and technological advances. Identifying and studying CTCs in the MRD setting could theoretically help us to identify genotypic and phenotypic metastatic subclones that could be targeted therapeutically and studied for resistance mechanisms. However, given the challenges in isolating CTCs in advanced disease settings, the development, validation and distribution of rapid, sensitive tests are important steps to increasing the success of routinely using CTCs in low tumour burden settings.
Conclusion
Following considerable scientific efforts to enhance genomic-profiling technologies, treatment strategies in GI cancers are increasingly relying on precision medicine. Whether it be through detecting MRD, risk-stratifying patients for adjuvant treatment strategies, guiding surveillance, characterizing baseline and emerging mutations, or predicting responses to therapy, ctDNA is a powerful tool with which we can individualize treatments for patients. While most data to date describe ctDNA in CRC, we are seeing the technology evolving across other GI cancers. Many questions regarding its interpretation in the MRD setting remain, but with emerging prospective studies and initiatives to incorporate biomarker analyses into clinical trials, we are hopeful the current uncertainties will be clarified in the future. We anticipate ctDNA will continue to be increasingly adopted in clinical practice to deliver personalized care and ultimately improve cancer outcomes.










