
Gastrointestinal stromal tumours (GISTs) are the most common mesenchymal tumours of the gastrointestinal (GI) tract, accounting for 0.1–3.0% of all GI malignancies, and 80% of all GI sarcomas.1–5 While GISTs can occur throughout the entire GI tract, with similar male/female incidence, 60% of cases usually present in the stomach, and about 30% in the small intestine.1 GISTs can occur at any age, but are most common between the ages of 40 and 60 years.6,7 GISTs that occur outside the GI tract are less frequent, and tend to develop in the omentum, mesentery and retroperitoneum.
The discovery that GISTs feature immunohistochemical expression of CD34 and KIT supports the hypothesis of origin from the interstitial cells of Cajal and differentiated GISTs from leiomyosarcoma, leiomyomas and gastric schwannomas. While it is known that GISTs arise from the same lineage as the interstitial cells of Cajal, it is not yet clear whether they arise from these cells, or their precursors.4,5 GISTs arise when genes responsible for the tyrosine kinase expression undergo a mutation that leads to a neoplastic growth involving the lineage of the interstitial cells of Cajal.8 When metastatic, the spread is mainly haematogenous, most commonly to the liver and peritoneum.1
The majority of GISTs – approximately 80% – are driven by a gain-of-function mutation in the KIT oncogene that encodes a 145-kD transmembrane receptor, KIT, with tyrosine kinase activity, leading to its activation in the absence of the ligand.9 About 5–10% of GISTs are driven by a mutually-exclusive activating mutation in platelet-derived growth factor receptor alpha (PDGFRα), leading to both downstream activation of signalling indistinguishable from KIT-mutated GISTs, and to the same uncontrolled proliferation and impaired apoptosis of these cells.10,11 While KIT and PDGFRα mutations may be sporadic, they may also be inherited, leading to much rarer familial GISTs.12 Rarely, there is a wild-type GIST that lacks mutation on KIT or PDGFRα, which compromise a heterogeneous group of mutations that include NF1, BRAF, HRAS, N-TRK, and SDH deficiency, and can be seen in tumour syndromes such as Carney triad, Carney–Stratakis syndrome and neurofibromatosis type.4,5,12,13
Approximately two decades have passed since the discovery of the most common driver mutation (KIT proto-oncogene), which led to the development of many active targeted therapies for GISTs. Since 2010, further findings have been achieved that extend our understanding beyond the inhibition of KIT and PDGFRα. Here, we aim to review the latest advances in the treatment of metastatic GIST and future perspectives.
Treatment
Surgical resection is the standard treatment for localized GISTs to achieve an R0 surgical outcome (optimal) with complete removal, leaving an intact capsule. However, the risk of local recurrence or metastatic disease may be as high as 40–50%, commonly spreading to the liver and/or abdominal cavity.14–17 GIST is an example of the successful development of target agents against critical driver alterations in cancer, and has led to a change in the outcome of this previously deemed ‘chemo-resistant’ tumour.
Tyrosine kinase inhibitors
The discoveries of KIT and PDGFRα mutations and their role in GIST biology have been essential for understanding the disease and its diagnosis, but most importantly, for treatment with tyrosine kinase inhibitors (TKIs). In 2001, the US Food and Drug Administration (FDA) first approved imatinib mesylate to treat malignant/metastatic and/or unresectable GIST. Before the approval of this drug, chemotherapy regimens had not yet been proven effective in treating GISTs, and median survival from time of diagnosis of metastatic or recurrent disease was reported to be 12–19 months.18,19
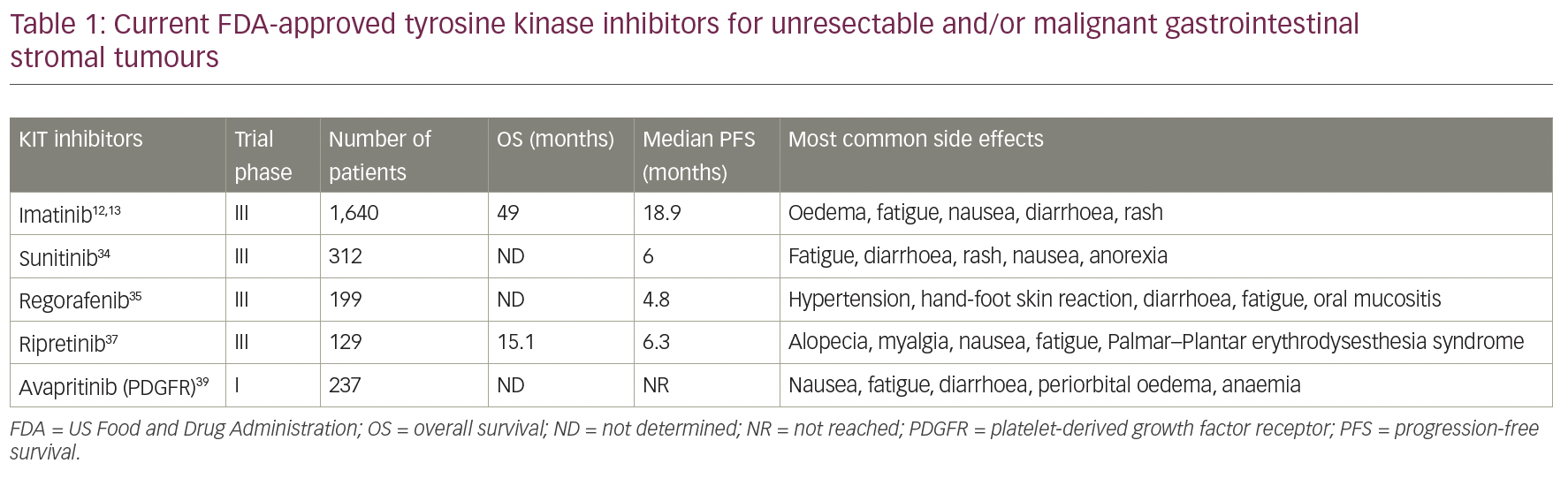
Imatinib mesylate is a protein-TKI that inhibits the receptor tyrosine kinase for platelet-derived growth factor and stem-cell factor, KIT, thereby reducing cellular proliferation and inducing apoptosis.10 It has been widely used for non-surgical management in the advanced/metastatic GIST setting since van Oosterom et al.,11 and shows significant activity in this group of patients. After this study, two open-label, randomized, multinational, phase III studies were conducted in patients with unresectable or metastatic malignant GIST.12,13 The two study designs were similar, allowing a predefined combined analysis of safety and efficacy. A total of 1,640 patients were enrolled into the two studies and randomized 1:1 to receive either 400 mg or 800 mg orally daily and continuously until disease progression or unacceptable toxicity. There was no significant difference among the doses with a median progression-free survival (PFS) of 18.9 months (95% confidence interval [CI] 17.4–21.2) and overall survival (OS) of 49.0 months (95% CI 45.3–60.0).12,13,20–22 Since then, imatinib continues to be recommended, not only as the standard first-line therapy for advanced GISTs, but also as adjuvant therapy for high-risk, localized GISTs by the European Society of Medical Oncology (ESMO), National Comprehensive Cancer Network (NCCN) Task Force, and the Japanese Society of Clinical Oncology (JSCO).7,23–25
Imatinib is currently recommended at a standard dose of 400 mg/day; the mutation type of the tumour impacts its response.26 Tumours harbouring KIT exon 11 mutations are more sensitive to imatinib, while GISTs with KIT exon 9 mutation experience a less favourable response unless treated at 800 mg/day. GISTs with PDGFRα exon 18 D842V mutations are resistant to imatinib.27–29 Despite this, imatinib at the 400 mg/day dose remains the standard initial therapy for metastatic GIST, and dose escalation to 800 mg/day is considered an option for patients progressing, particularly in KIT exon 9 mutant.
Although imatinib significantly improves both PFS and OS in patients with advanced GISTs, most patients treated with imatinib will eventually develop resistance to it, either primarily (within 180 days of therapy) or secondarily (after 180 days of therapy).30–32 Imatinib-resistant GISTs continue to depend on KIT signalling for cell survival and proliferation, as well as on the PI3K/mammalian target of rapamycin, the RAS/RAF/MEK/MAPK signalling pathways, and the inhibition of KIT downstream pathway, which remains a possible target in these tumours. With this in mind, there are currently four additional drugs that act on this same pathway and are FDA approved for GIST treatment, with imatinib remaining as the first-line treatment.
Sunitinib, an oral multitarget TKI with activity against KIT, PDGFRα, and vascular endothelial growth factor receptor (VEGFR), was the second FDA-approved drug to treat resistant/metastatic GIST, and is considered second-line therapy for these patients. While shown to be useful for exon 13/14 mutations involving the ATP binding site, like imatinib, it has poor activity for the mutations affecting the activation loop, such as exon 17/18.33 Its efficacy was demonstrated in a clinical trial in 2003 (ClinicalTrials.gov Identifier: NCT00075218) that studied 312 patients with advanced disease and resistance or intolerance to imatinib. Patients were randomized 2:1 to sunitinib or placebo. PFS was 24.1 weeks for sunitinib (95% CI 11.1–28.3) versus 6.0 weeks for placebo (95% CI 4.4–9.9), and therapy was well tolerated.34 Unfortunately, clinical progression and drug resistance to sunitinib subsequently evolve, generally within 1 year of treatment.35
Regorafenib is a third-line, FDA-approved therapy, which was approved in 2013 and acts on KIT, PDGFR, VEGFR and BRAF pathways. Its efficacy was demonstrated in a randomized clinical trial involving 57 hospitals in 17 countries, with a total of 199 patients required to have progressed on, or be intolerant to, imatinib and sunitinib therapies. Patients were randomized to regorafenib or placebo, and results showed a median PFS of 4.8 months versus 0.9 months, respectively, as well as a disease control rate of 52.6% versus 9.1%, respectively. The most common adverse events reported were hypertension, hand-foot skin reaction, and diarrhoea.35 Moreover, a phase II trial (ClinicalTrials.gov Identifier: NCT02606097) showed that regorafenib might have a PFS benefit when it is used upfront for patients with GISTs harbouring exon 17 mutations with stable disease compared with those experiencing disease progression (median PFS: not reached versus 12.9 months). Thus, in this specific population, regorafenib might be used irrespective of imatinib or sunitinib use.36
Recently, two new TKIs were approved by the FDA for patients with GIST, in which imatinib and sunitinib have failed. Ripretinib, a switch-control TKI that broadly inhibits KIT and PDGFRα kinase signalling through a dual mechanism of action, was approved in January 2020 for patients who progressed on three or more kinase inhibitors, including imatinib. Efficacy was evaluated in the INVICTUS trial (ClinicalTrials.gov Identifier: NCT03353753) – a controlled study in 129 patients with GIST previously treated with imatinib, sunitinib and regorafenib. Patients received 150 mg. of ripretinib or placebo, daily until disease progression or unacceptable toxicity. Median PFS was 6.3 months (95% CI 4.6–6.9) with ripretinib, compared with 1.0 month (95% CI 0.9–1.7) with placebo (hazard ratio 0.15, 95% CI 0.09–0.25; p<0.0001).37 Ripretinib was later evaluated in a phase I study with dose escalation from a daily dose to twice daily (ClinicalTrials.gov Identifier: NCT02571036). This was presented at ESMO 2020, demonstrating a PFS clinical benefit across all treatment lines, with a similar safety profile.38
Despite the promising findings in TKIs, none of them target PDGFRα Asp842Val (D842V), the primary driver mutation in 5–6% of GISTs. Avapritinib was evaluated in the phase I clinical trial NAVIGATOR (ClinicalTrials.gov Identifier: NCT02508532), which included patients with unresectable PDGFRα D842V-mutant GIST regardless of previous therapy or presence of other mutations, that either progressed on imatinib and one or more TKI, or only received imatinib previously. It showed high activity and safety in both groups of patients, and in February 2020, the FDA approved the use of avapritinib for the treatment of adults with unresectable or metastatic GIST harbouring a PDGFRα exon 18 mutation, including PDGFRα D842V mutations.39 A phase III trial, VOYAGER, compared avapritinib with regorafenib in the third-line setting for patients with locally advanced unresectable or metastatic GIST, and observed a superior objective response rate with avapritinib, but found no difference in the primary endpoint of median PFS.40 Table 1 reflects all current FDA-approved TKIs for unresectable and/or malignant gastrointestinal stromal tumours mentioned.
Novel tyrosine kinase inhibitors and clinical trials
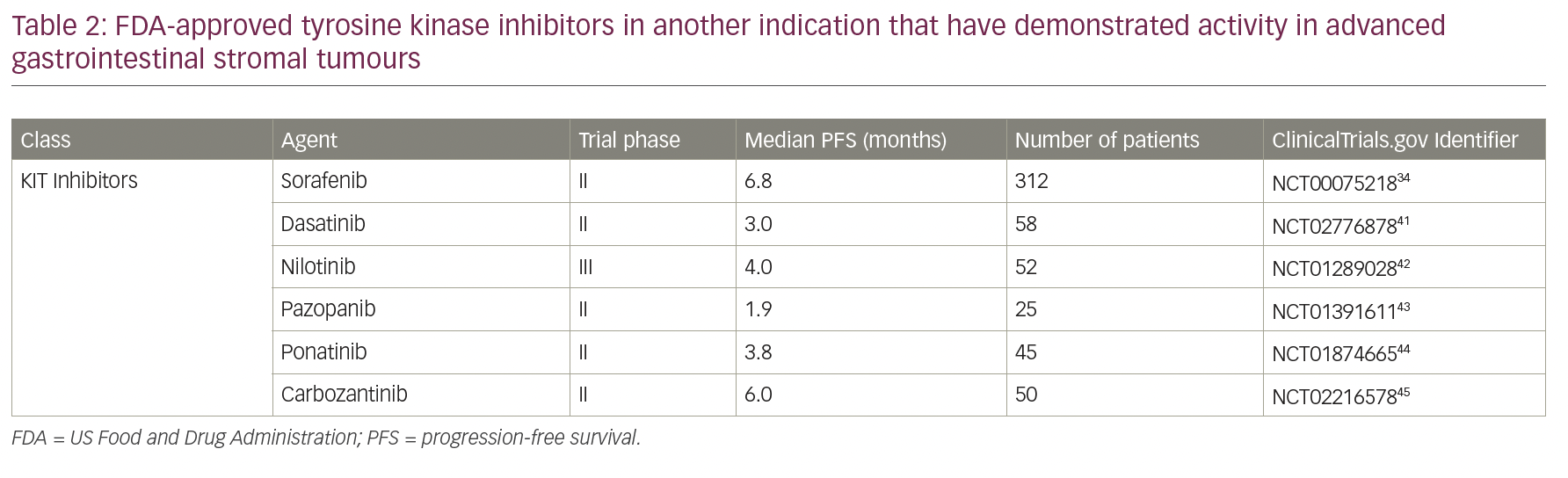
Despite the current treatment options for resistant/metastatic GIST, there are still unmet needs. Multiple other TKIs that are approved for different diseases have been tested in GISTs, but remain non-FDA approved and can be used only as off-label. These options are especially relevant in cases when no other available treatment is available and/or participating in a clinical trial is not feasible for the patient (Table 2).32,41–45
Another possible target within the KIT downstream pathway is the transcription factor ETV1, a regulator of the interstitial cells of Cajal that plays a major role in GIST development. Dual inhibition of KIT and ETV1 by imatinib and the MEK inhibitor MEK162 has been reported in the literature both in vitro and in vivo.46 A phase II study is currently exploring the effect of imatinib in combination with MEK162 in patients with untreated advanced GIST (ClinicalTrials.gov Identifier: NCT01991379). In 2019, Rosenbaum et al. published a phase I study on the combination of a MEK162 (binimetinib) with pexidartinib (a potent inhibitor of CSF1R, KIT, and FLT3) in patients with advanced or metastatic GIST who progressed on imatinib; it was found to be tolerable and had PFS and OS of 6.1 and 14.6 months, respectively (ClinicalTrials.gov Identifier: NCT03158103).47 More recently, a phase II study of binimetinib in combination with imatinib in patients with untreated advanced GIST was presented at ASCO 2020 (ClinicalTrials.gov Identifier: NCT01991379), with a primary endpoint of RECIST 1.1 objective response rate. The primary endpoint was met, indicating that this combination is highly effective in treatment-naïve advanced GIST.48 Additionally, at the last Connective Tissue Oncology Society Annual Conference, Trent et al. presented the result of a phase I trial that evaluated a novel TKI, PLX9486, combined with another target drug, such as sunitinib, and showed how this combination could be feasible and tolerable in the majority of patients. They reported a clinical benefit, with a median PFS of 12 months in the PLX9486-naïve group, and 11 months for the entire group of 18 patients involved in the trial.49
Other than drugs being developed in the KIT downstream pathway, several multi-kinase inhibitors have been studied with potent activity against GIST. Dovitinib, an inhibitor of VEGFR 1–3, fibroblast growth factor receptors 1–3, PDGFRβ and KIT, was studied in phase II trials. In the first trial, patients with GIST refractory to imatinib and sunitinib were included, and dovitinib was found to have activity with a median PFS of 3.6 months; in the second trial, patients who progressed on imatinib were analysed, and the median PFS was 4.6 months.50,51 Valatinib, a VEGFR, KIT and PDGFR inhibitor, was also tested in a phase II trial in patients with GIST who were resistant to the first and second lines of treatment, and was approved by the FDA, with a median time to progression of 5.8 months when used as a second-line therapy, and 3.2 months when as a third-line therapy.52
For PDGFRα mutant tumours, crenolanib – an orally bioavailable benzimidazole – was shown to have activity against PDGFRα D842V mutant GIST in a phase II trial (ClinicalTrials.gov Identifier: NCT01243346). Currently, a phase III randomized, placebo-controlled trial of oral crenolanib versus oral placebo in combination with best supportive care in patients with advanced or metastatic GIST with a D842V mutation in the PDGFRα, is ongoing and actively recruiting patients (ClinicalTrials.gov Identifier: NCT02847429).
The role of immunotherapy
Over the past decade, we have seen a revolution in cancer treatments by moving away from standard therapies (chemotherapy and radiation) to the use of antibody-based immunotherapies that modulate immune responses against tumours, based on the idea that these standard therapies may enhance immune activity. Tumour-infiltrating immune cells, which include CD8+ T cells (cytotoxic) and regulatory T cells (suppressive cells), create independent immunosuppressive networks and play a role in tumour surveillance and progression. The balance between these is regulated by cytokines, checkpoint proteins and other types of immune cells.53 The first generation of antibody-based immunotherapy, so-called immune-checkpoint blockade, works by blocking the receptor and/or ligand interactions of molecules, such as cytotoxic T-lymphocyte antigen 4 (CTLA-4) and programmed cell death protein 1 (PD-1), that are involved in diminishing T-cell activation or function.54
The microenvironment of GISTs is populated by tumour-infiltrating immune cells; this microenvironment is important to enhance the antitumor function of imatinib, as TKIs can manipulate the immune system. In preclinical studies on KIT mutant GIST mouse models, imatinib was shown to have an antitumor effect by directly inhibiting tyrosine kinases on tumour cells, and indirectly through the immune system when activating CD8+ cells and inducing regulatory T-cell apoptosis.55
Checkpoint inhibitors are not yet approved in the treatment of patients with GIST, but several clinical trials have investigated the efficacy of immunotherapy in this tumour. Table 3 illustrates ongoing/completed trials that aimed to evaluate the response of immunotherapy in GIST.56,57 D’Angelo et al. presented a phase Ib trial with the combined KIT and CTLA-4 blockade in eight patients with refractory GIST and showed few durable Choi responses.58 In 2018, a phase Ib/II trial assessed the efficacy and safety of an anti-PD-1 antibody, named spartalizumab (PDR001), plus imatinib. The trial included patients with metastatic or unresectable GIST with prior failure on imatinib, sunitinib or regorafenib.59 The combination of immunotherapy and a second agent (non-immunotherapy) in the management of advanced GIST is the subject of several clinical trials: imatinib plus ipilimumab (phase I); axitinib (anti-VEGFR/KIT/PDGFR) plus pembrolizumab (phase II); metronomic cyclophosphamide plus pembrolizumab (phase II); spartalizumab (PDR001) plus imatinib (phase Ib/II); and PLX3397 (TKI) plus pembrolizumab (phase I/IIa), which had to be terminated early due to insufficient evidence of clinical efficacy.57,60
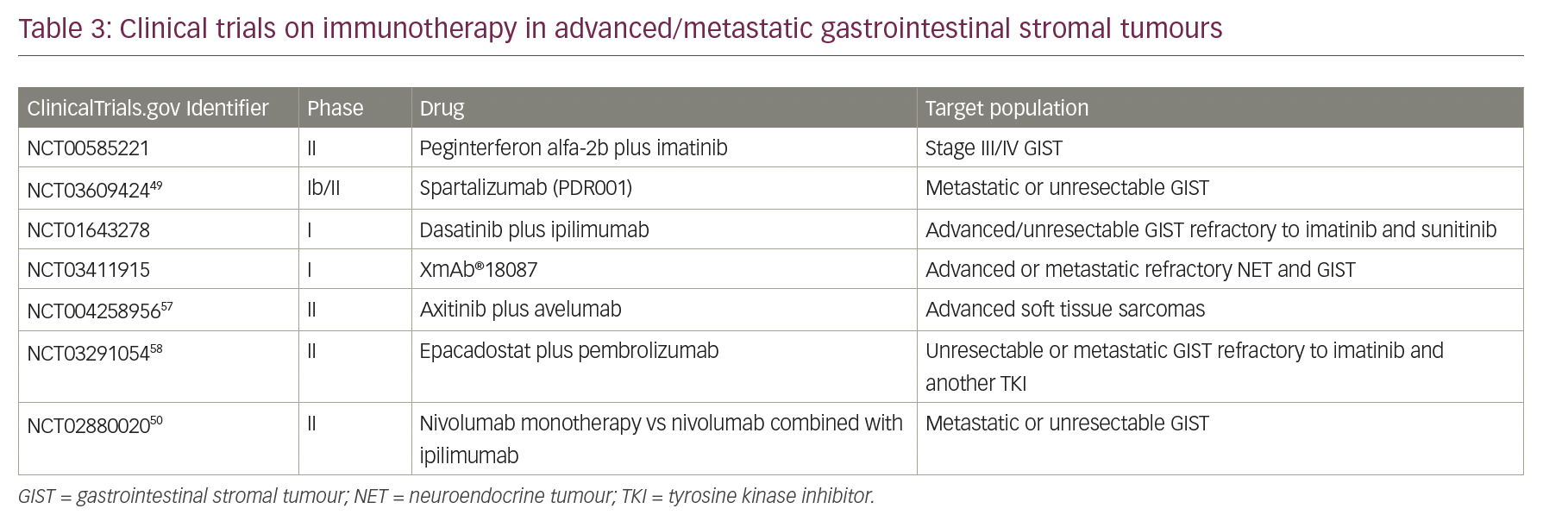
Dual blockade of two different checkpoint proteins with PD-1 and CTL-4 inhibitors is also being studied in the advanced GIST population, but with some contrasting reports. While the phase II study of nivolumab monotherapy versus nivolumab combined with ipilimumab, presented at ASCO 2020 reported benefits in both response and disease control in heavily pre-treated patients with GIST (ClinicalTrials.gov Identifier: NCT02880020),57 the results of the Alliance A091401 cohort expansion of the phase II trial of nivolumab +/- ipilimumab for patients with metastatic sarcoma reported that neither leads to confirmed response rates in GIST.62
Cellular therapy and GIST are also being studied, but are still in the preclinical setting. Katz et al. reported that chimeric antigen receptor T cells, produced with anti-KIT activity that binds GIST cells, produces interferon-gamma and lyse the cells in vitro, resulting in a significant reduction in tumour growth.63
Conclusion
Major advances have been made in the management of GIST over the past 20 years, since the discovery of the first TKI to now five different FDA-approved agents for the treatment of advanced and metastatic GIST. Since most patients eventually become refractory to available agents, there remains an unmet therapeutic need. Genome editing technologies have tremendous potential in sarcoma biology research and development of therapies, as sarcomas are believed to develop from genetic alterations in mesenchymal progenitor cells, offering exciting opportunities for the future. Patients with GIST should be consulted and referred to sarcoma expert centres for disease management and treatment due to the several nuances and novelties that are involved in their treatment.










