Cholangiocarcinomas (CCAs) are a heterogenous group of cancers arising from the biliary epithelium. While they account for around 3% of all gastrointestinal malignancies, they are the second most common primary liver cancer.1 CCAs tend to metastasize and have early lymph node involvement. As a result, only a small proportion of patients are considered for surgical management, which offers the only curative treatment. In very select patients, liver transplantation may be considered.2 CCAs are subdivided based on the anatomical origin: intrahepatic CCA (iCCA) arises from the small ducts within the periphery of the liver; perihilar CCA (pCCA) arises at the level or junction of the right and/or left hepatic ducts; and distal CCA (dCCA) usually involves the common bile duct.3,4 The anatomic distinction between pCCA and dCCA occurs at the level of the cystic duct.3,4 Together, pCCA and dCCA account for 70–80% of all biliary tract cancers, while iCCA accounts for 10–20%.1,3,5,6 Recently, the incidence of iCCA has been rising.7 The anatomic variations align with the differences seen in molecular targets in these subsets, and these can help stratify this heterogenous disease. Given the paucity of successful therapies in these cancers, it is important to understand the molecular drivers and to develop therapeutic strategies using targeted therapies either alone or in combination.
Patterns of molecular targets
Molecular-based therapies have been successful in treating many types of malignancies and, in some cases, have become the standard over traditional chemotherapy. However, developing targeted therapies in CCAs is challenging, in part due to the inter- and intra-tumoural heterogeneity of the disease. The genetics and molecular mechanism of early and advanced disease remain somewhat elusive despite the strides made in molecular testing. In the era of mutational testing with next generation sequencing, the molecular landscape has demonstrated several new potentially targetable drivers (Figure 1). Genomic diversity according to the underlying aetiological risk factors is illustrated by the higher mutational burden found in liver fluke-driven tumours and the preponderance of erb-B2 receptor tyrosine kinase 2 (ERBB2, also known as human epidermal growth factor 2 [HER2]) amplifications and tumour protein p53 (TP53) mutations.8 Conversely, non-liver fluke CCAs harbour high copy-number aberrations, programmed death 1 (PD-1) and programmed death-ligand 1 (PD-L1) expression, epigenetic mutations involving isocitrate dehydrogenase 1/2 (IDH1/2) and breast cancer gene 1-associated protein-1 (BAP1), and fibroblast growth factor receptor (FGFR) rearrangements.8,9

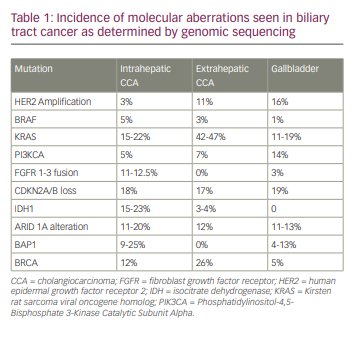
Recently, Nakamura and colleagues conducted comprehensive whole-exome and transcriptome sequencing in 260 patients with biliary tract cancers, which included 145 patients with iCCA, 86 with pCCA/dCCA and 29 with gallbladder cancer.10 They demonstrated that a potentially targetable genetic alteration was seen in about 40% of patients.10 Additionally, IDH1, IDH2, FGFR1, FGFR2, FGFR3, ephrin type-A receptor 2 (EPHA2) and BAP1 mutations were recurrent in iCCAs, while AT-rich interaction domain 1B (ARID1B), E74-like factor 3 (ELF3), polybromo 1 (PBRM1), protein kinase cyclic adenosine monophosphate-activated catalytic subunit α (PRKACA) and PRKACB mutations occurred preferentially in pCCA/dCCA.10 Additional genomic sequencing studies have revealed that 30–40% of iCCAs have actionable mutations, which include IDH1/2 (5–20%), FGFR2 fusions (4–16%), ARID1A alterations (7–16%) and BAP1 mutations (1–38%).11 iCCAs commonly have mutations in IDH1/2 and BAP1, and FGFR fusions, and extrahepatic CCAs carry Kirsten rat sarcoma virus (KRAS), TP53 and mothers against decapentaplegic homologue 4 (SMAD4) mutations more commonly.12,13 Conversely, gallbladder cancers harbour a high frequency of human epidermal growth factor receptor 2 (HER2) mutations.12,13 A summary of the molecular aberrations seen in BTCs are shown in Table 1.
Molecular targeted therapy in intrahepatic cholangiocarcinoma
Fibroblast growth factor receptor pathway
The FGFR family consists of four subtypes of transmembrane tyrosine kinase receptors, FGFR1–4.14 FGFR signalling plays an essential role in several cellular processes, including proliferation, survival and migration, and mediates several vital physiological functions, such as metabolic homeostasis, neuroendocrine balance, embryonic development and tissue repair.15,16 Dysregulation precedes malignant transformation and presents as gene amplification, gain-of-function coding mutation and gene fusion.15,16 Resulting from this cascade, there is downstream activation of FGFR substrate 2 (FRS2), mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase 1/2 (ERK1/2), Janus kinase–signal transducer and activator of transcription (JAK-STAT), phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signalling pathways.15–18 In turn, intracellular phosphorylation of receptor kinase domains, cascading reactions to intracellular signals and gene transcription are activated.16,17
iCCAs more commonly express FGFR mutations compared with other mutations, with approximately 13–14% having FGFR2 fusions/translocations, which may be mutually exclusive with KRAS mutations.19 The most common FGFR chromosomal aberration seen in CCA is the constitutively active FGFR2–BicC family RNA binding protein 1 (BICC1) fusion, which has a role in activating the MAPK and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit α (PIK3CA) and mechanistic target of rapamycin (mTOR) pathways.16,20 In vitro and in vivo studies have shown that overexpression of FGFR2 fusion proteins is sensitive to FGFR inhibition.21 Several FGFR inhibitors have been evaluated for clinical benefit and, recently, the US Food and Drug Administration (FDA) approved two FGFR2 inhibitors, infigratinib and pemigatinib in several cancers including CCAs in patients harbouring these mutations.22,23
FIGHT-202 was a multicentre, open-label, single-arm phase II study evaluating pemigatinib, a selective and potent oral inhibitor of FGFR1, 2 and 3.24 Included were patients with previously treated, locally advanced or metastatic CCA with or without FGFR2 fusions or rearrangements. In this study, patients were assigned to one of three treatment arms: FGFR2 fusions or rearrangements, other fibroblast growth factor (FGF)/FGFR alterations, or no FGF/FGFR alterations. Overall, 146 patients were enrolled: 107 with FGFR2 fusions or rearrangements, 20 with other FGF/FGFR alterations, 18 with no FGF/FGFR alterations and 1 with an undetermined FGF/FGFR alteration. Among the 107 patients with FGFR2 fusions or rearrangements, the overall response rate (ORR) was 36% (95% confidence interval [CI] 27–45), which included three complete responses. The median duration of response was 9.1 months, with 24/38 (63%) of patients having a response for more than 6 months and 7/38 (18%) patients having a response for more than 12 months. The median progression-free survival (PFS) was 6.9 months (95% CI 6.2–9.6) and most patients (61%) received one previous line of systemic therapy. Around 60% of patients on the trial experienced hyperphosphataemia, the most common adverse event.24 Based on these results, the FDA granted accelerated approval in April 2020.25 These results formed the premise for the FIGHT-302 trial, a randomized, open-label phase III study that will evaluate the efficacy and safety of first-line pemigatinib versus gemcitabine and cisplatin in unresectable/metastatic CCA with FGFR2 fusions or rearrangements (ClinicalTrials.gov identifier: NCT0365636).26 The study is currently recruiting, and its primary endpoint is PFS.26
In early 2021, the FDA granted accelerated approval to infigratinib, an adenosine triphosphate-competitive FGFR1–3 oral tyrosine kinase inhibitor.27 This was evaluated in a phase II study with a planned enrolment in pre-treated patients with advanced CCA, divided into three cohorts: 120 patients with FGFR2 gene fusions or rearrangements; 20 patients with FGFR1 and FGFR3 fusions or rearrangements and/or FGFR mutations; and 20 patients with FGFR2 gene fusions who have disease progression after prior treatment with a selective FGFR inhibitor other than infigratinib.27 In cohort 1 (patients with FGFR2 gene fusions or rearrangements without receiving a prior FGFR inhibitor), 108 patients were enrolled. ORR was 23.1% (95% CI 15.6–32.2) and included one patient with a complete response and 24 patients with partial response. Median duration of response was 5.0 months (range 0.9–19.1 months). Of the patients who responded to treatment, 8 (32%) patients had a response lasting >6 months, and median PFS was 7.3 months (95% CI 5.6–7.6 months).27 Interestingly, the ORR was 34% (17/50) in patients who received infigratinib in the second-line setting. The most common treatment-related adverse events included hyperphosphataemia (76.9%), eye disorders (67.6%, excluding central serous retinopathy), stomatitis (54.6%) and fatigue (39.8%).28 PROOF, a phase III study of infigratinib versus gemcitabine and cisplatin is on-going in the front-line setting (ClinicalTrials.gov identifier: NCT03773302).29
Similarly, futibatinib (TAS120), a highly selective, irreversible, FGFR1–4, oral tyrosine kinase inhibitor, inhibits cancer cell growth in human xenografts of tumours bearing FGFR aberrations.30 In a phase II trial of 103 patients with FGFR2 gene fusions, the confirmed objective response rate was 41.7% (43/103).31 Responses were durable, with a median duration of response of 9.7 months, with 72% of responses lasting longer than 6 months. The disease control rate (DCR) was 82.5%, and the median PFS and median overall survival (OS) were 9.0 months and 21.7 months, respectively.31 To add to these results, there is also an on-going phase III trial comparing futibatinib with gemcitabine and cisplatin as first-line therapy in patients harbouring FGFR2 aberrations.32
One of the concerns with first-generation FGFR2 inhibitors (pemigatinib and infigratinib) is the emergence of resistance. A study showed recurrent point mutations at the FGFR2 kinase domain upon progression in serial cell-free DNA (cfDNA) specimens from patients who had progressed on infigratinib.33 Furthermore, post-progression lesion biopsies showed marked inter- and intra-lesional heterogeneity with different FGFR2 mutations in individual resistant clones.33 Hence, polyclonal secondary FGFR2 mutations appear to represent an important clinical resistance mechanism. More recently, in patients who had progressed on infigratinib, targeted sequencing of tumour DNA revealed FGFR2 kinase domain p.E565A and p.L617M single-nucleotide variants, which are potential drivers of the resistance.34 Further proteomics identified upregulation of the PI3K/AKT/mTOR signalling pathway in cells harbouring the FGFR2 p.E565A mutation, suggesting that combining an FGFR2 inhibitor and an mTOR inhibitor could be an approach for patients with infigratinib resistance.34 In future studies targeting FGFR2, combination therapies may address these potential resistance mechanisms.
Isocitrate dehydrogenase 1
IDH is an essential metabolic enzyme for cellular respiration in the tricarboxylic acid cycle. IDH1 and IDH2 catalyse the nicotinamide adenine dinucleotide phosphate (NADP)-dependent oxidative decarboxylation of isocitrate to α-ketoglutarate and CO2.35 The gain-of-function IDH mutation disrupts the normal catalytic activity of IDH1/2, leading to increased conversion of α-ketoglutarate to D-2-hydroxyglutarate (D-2HG).8,36 The D-2HG acts as an onco-metabolite to increase cell proliferation, potentiate metastases and activate pathways such as vascular endothelial growth factor (VEGF).36
IDH mutations occur in about 15–20% of all iCCAs, with the common allelic variations being IDH1-R132C and IDH1-R132G.10,36 In general, IDH1 mutations tend to be mutually exclusive of KRAS/neuroblastoma rat sarcoma virus viral oncogene homologue (NRAS) and FGFR mutations but can co-occur with BAP1.8,34
Phase I results from ivosidenib, the first orally available IDH1 inhibitor, showed that it was well tolerated, had clinical activity and reduced intra-tumoural 2-HG levels.37 This was the premise for the recent phase III ClarIDHy trial, where 230 patients with advanced, pre-treated, IDH1-mutated CCA were randomized 2:1 to ivosidenib or a placebo.38 Crossover was permitted at progression if a patient was in the placebo arm. Ivosidenib had a superior median PFS compared with placebo (2.7 months versus 1.4 months, respectively; hazard ratio [HR] 0.37; one-sided p<0.0001).38 Ivosidenib recently received FDA approval in this patient population.39 Given these promising results, a combination trial of ivosidenib with gemcitabine and cisplatin-based chemotherapy is on-going (ClinicalTrials.gov identifier: NCT04088188).40
There are preclinical data to support the hypothesis that IDH1 mutations are also dependent on SRC signalling.41 As such, SRC inhibitors (such as dasatinib) may augment the response of IDH1 inhibitors when combined, and a phase II trial evaluating this has just completed (ClinicalTrials.gov identifier: NCT02428855).42
Human epidermal growth factor receptor 2
The HER family of receptors comprises four receptors (HER1, HER2, HER3, HER4), all of which have an extracellular ErbB ligand-binding domain, a single transmembrane lipophilic segment, an intracellular tyrosine kinase domain and an intracellular C-terminal tail.43 The HER2 receptor is dimerized, which activates downstream signalling cascades, including the PI3K/protein kinase B (PKB or Akt) pro-survival pathway and the MAPK proliferation pathway.43,44 Mutational analysis of 75 surgical specimens with next-generation sequencing classified ERBB2 genetic aberrations in 20% of pCCAs/dCCAs and only 1.8% of iCCAs.13
Recently, MyPathway, a basket trial, evaluated the efficacy of dual HER2 therapies in HER2-positive, metastatic biliary cancer.45 Patients received pertuzumab (a monoclonal antibody that binds to domain II of HER2, inhibiting ligand-induced HER2/3 dimerization) + trastuzumab (a monoclonal antibody that binds to domain IV). At data cut-off, median follow-up was 8.1 months. Partial response was seen in 9 patients (23%, 95% CI 11–39%). An additional 11 patients (28%) had stable disease for 4 months or longer, yielding a DCR of 51%. Median PFS was 4.0 months (95% CI 1.8–5.7 months). The median OS was 10.9 months (95% CI 5.2–15.6 months).45 These results are promising for larger randomized trials of pertuzumab + trastuzumab in this previously treated population with advanced disease. Several other clinical trials investigating newer HER2 therapies are on-going, including with zanidatamab (ClinicalTrials.gov identifier: NCT04466891),46 GQ1001 (an antibody–drug conjugate against HER2; ClinicalTrials.gov identifier: NCT04450732)47 and tucatinib + trastuzumab (ClinicalTrials.gov identifier: NCT04579380).48
Proto-oncogene B-Raf mutations
Proto-oncogene B-Raf (BRAF) mutations encode protein kinases downstream of RAS as part of the MAPK signalling cascade. In turn, these promote cell proliferation, survival and malignant transformation.49,50 BRAF mutations are reported in up to 6% of all biliary cancers; and further enriched in up to 22% of iCCAs.49,50 Patients with BRAF mutations typically have poor prognosis, worse OS, higher tumour grade and chemotherapy resistance.51 Single-agent BRAF inhibitors, while promising, do not induce durable response as seen in other gastrointestinal cancers.52 A phase II trial of dabrafenib plus trametinib in BRAF-mutated rare gastrointestinal cancers demonstrated an overall response rate of 41% (13 of 32 patients) in the cohort with biliary tract cancers. Median PFS and median OS were 7.2 months and 11.3 months, respectively.53
Chromatin remodelling genes and histone deacetylase inhibitors
Chromatin remodelling is the process by which genomic DNA accesses regulatory transcriptional proteins and thereby controls gene expression. The primary genes involved in this function are ARID, BAP1, BRM and mixed-lineage leukaemia (MLL).11,54 These are often mutated, which drives carcinogenesis in several cancer types, including CCAs, uveal melanoma and mesotheliomas.54 Encoding nuclear deubiquitinase, BAP1 is involved in chromatin remodelling, while subunits of the switch/sucrose non-fermentable (SWI/SNF) chromatin remodelling complexes are encoded by ARID1A and PBRM1.11,54 These mutations are associated with poor outcomes across all cancer types.11 While there are no direct inhibitors of these mutations, there are several small molecules that target this chromatin remodelling, including histone deacetylase inhibitors vorinostat, romidepsin and azacytidine.3
Molecular targets in distal cholangiocarcinoma
There is a paucity of data defining the molecular landscape of dCCAs and there is, so far, no targeted therapy for these cancers. Recently, Montal et al. performed genomic analysis on 181 samples of extrahepatic CCA and found that KRAS (36.7%), TP53 (34.7%), ARID1A (14.0%) and SMAD4 (10.7%) were the most prevalent mutations.55 Additionally, they identified four clusters: metabolic class with hepatocyte-like features and expression of hepatocyte nuclear factor 4 (HNF4); proliferation class (23%), characterized by enrichment of Myc targets, ERBB2 mutations or amplifications, and activation of mTOR signalling; mesenchymal signalling, which had poor prognosis and aberrant transforming growth factor (TGF)-β signalling; and immune class (11%), which had increased lymphocyte infiltration, overexpression of PD-1/PD-L1 and molecular features corresponding to improved immune checkpoint inhibition.55,56 The genomic landscape of each class provides the foundation for exploring patient stratification and novel therapeutic approaches. In addition, dCCAs are also associated with ELF3 mutations.57 These appear to also drive ampullary cancers, and to date, no therapeutic drug is available for this.57
Molecular targets across all subtypes
Kirsten rat sarcoma virus
The proto-oncogene KRAS is one of the most prevalent mutations in CCAs.13 Historically, patients with KRAS have poor prognoses and shorter OS.13 Until recently, there has been no therapeutic approach to target KRAS. Sotorasib is a small molecule that selectively and irreversibly targets KRASG12C, and was recently approved by the FDA for patients with the mutation in non-small-cell lung cancer.58,59 However, KRASG12C is rare in biliary cancer and is only seen in about 1.8% of patients.60 KRASG12C inhibitors are being investigated in other cancer types (Clinical trials.gov identifier: NCT03600883), its utility in BTC remains to be seen.59,61
Mesothelin
Mesothelin is a glycosylphosphatidylinositol (GPI)-anchored membrane protein that is a promising target for antibody-directed cancer therapy. In healthy individuals, mesothelin is present in mesothelial cells of the pleura, peritoneum and pericardium at relatively low levels; however, it is highly expressed in several different cancers, including pancreatic cancers, CCAs and breast cancer.62 In a retrospective study of tissue from 61 patients with extrahepatic CCA, mesothelin expression was evaluated.63 High expression correlated with poor prognosis, metastatic disease and peritoneal carcinomatosis.63,64
DNA damage repair genes
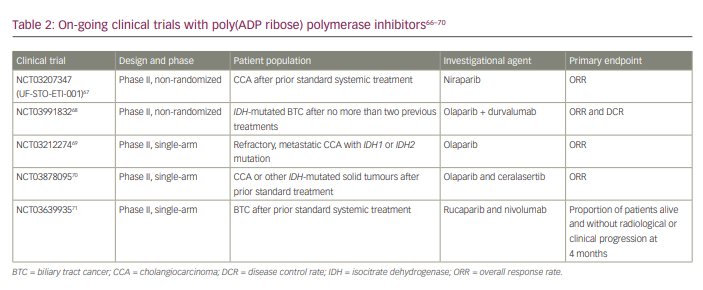
DNA damage repair (DDR) is a crucial component of genomic stability. Both germline and somatic mutations in these genes increase the risk of cancers. Abnormal DNA repair genes increase mutations and alterations, resulting in genetic mistakes accumulating.65 Ross et al. profiled 321 patients with biliary tract cancer for DNA repair genes (mutS homologue 6 [MSH6], MSH2, breast cancer type 1 [BRCA1], BRCA2, ataxia-telangiectasia mutated [ATM] or mutL homologue 1 [MLH1] genes). They found DDR mutations in 12% of patients with iCCA, 26% of patients with extrahepatic CCA and 5% of patients with gallbladder cancer.65 Poly(ADP ribose) polymerase (PARP) inhibitors are oral small-molecule inhibitors of PARP1, PARP2 and PARP3, whose actions are based on synthetic lethality. Inhibiting PARP causes the persistence of single-strand breaks, ultimately resulting in double-strand breaks.66 There are several trials currently ongoing looking at the combination of PARP inhibitors and immunotherapy (Table 2).67–71
Neurotrophic tropomyosin receptor kinase gene fusions
Neurotrophic tropomyosin receptor kinase (NTRK) fusions, specifically NTRK1, NTRK2 and NTRK3 gene fusions, have oncogenic driver properties in many cancers, including CCAs. The molecular aberrations occur with the fusion of the C-terminal tyrosine kinase with a N-terminal fusion partner, thereby leading to ligand-independent phosphorylation and cell proliferation.72 These fusions are very rare, and a recent study observed a prevalence of less than 1% in patients with CCA.73
Tropomyosin receptor kinase inhibitors entrectinib (RXDX-101) and larotrectinib (LOXO-101), have recently been approved in NTRK-fusion-positive malignancies.74 In basket trials, tumour-agnostic therapies have shown excellent activity in previously treated patients with an NTRK fusion, with ORRs of 55–75%.72,75 Recent combined results from the STARTRK-1, STARTRK-2 and ALKA-372-001 trials suggest meaningful response with entrectinib in gastrointestinal cancers, including CCAs, with a tolerable safety profile; 1 patient achieved a partial response and PFS of 12 months.76
Immunotherapy for cholangiocarcinoma
In cancer, immune checkpoints are dysregulated and drive immune resistance and immune escape.77 There are several reasons for immune resistance, including altered tumour microenvironment, which creates an immunosuppressive setting, expression of immune checkpoint proteins such as PD-1 and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and loss of major histocompatibility complex expression.3,78 Recognizing that checkpoint molecules are co-inhibitory receptors of T-cell activation was paradigm shifting, and led to the notion that blocking anti-tumour T-cell responses and removing these signals would have an anti-tumour effect.3,78,79
Nakamura et al. reported that in 260 patients with biliary tract cancers, the expression of nine targetable immunosuppressive immune checkpoint molecules, including PD-L1 were seen in 45.2% of cases.10 Correspondingly, treatment with an antibody to PD-L1 elicited a favourable clinical response.10 Furthermore, in immunohistochemical staining of surgical specimens of patients with iCCA, tumours expressed more PD-1 in iCCA tissue compared with adjacent tissue.3,80 The authors also demonstrated that tumour-associated PD-L1 expression correlated with worse tumour stage and poorly differentiated tumours. As well, higher expression of PD-L1 inversely correlated with CD8+ tumour-infiltrating lymphocytes (TILs), but not CD4+ TILs.80
These findings beg the question of clinical responses with immunotherapy in these patients. Recently, Carapeto et al. evaluated the immune profile of 96 patients with iCCA with resected tumours.81 The authors strategically combined immune checkpoint immunohistochemistry with next generation sequencing. They demonstrated that T-cell and immune-checkpoint markers were expressed differently at the tumour margins compared with the tumour centre. lymphocyte-activation gene 3 (LAG3) and low CD3/CD4 at the tumour centre. Interestingly, increased expression of the immunosuppressive checkpoint marker B7H4 was linked to BAP1 mutations.81 Several trials are on-going evaluating the different immunotherapy agents available.
Response to treatment with PD-1 or PD-L1 immune checkpoint inhibitors correlates with a high level of microsatellite instability (MSI-H).82 However, data about the prevalence of MSI-H in CCA remain limited. In a small study of 4 patients with biliary tract cancers who had MSI-H and were treated with pembrolizumab, an anti-PD-1 monoclonal antibody, the DCR was 100%. One patient achieved a complete response and three patients had stable disease.82
The clinical evidence for the use of immunotherapy targets in patients with microsatellite-stable CCAs is limited. The safety and efficacy of pembrolizumab were evaluated in the basket trial, KEYNOTE-028 across several different cancer types.83 Thirty-seven patients with PD-L1-positive biliary tract cancers were included in the trial. The definition of PD-L1 positivity was staining in ≥1% of cells in tumour nests or PD-L1-positive bands in stroma by immunohistochemical assay. The duration of response was 5.4–9.3 months, with an overall response rate of 17%. Stable disease was seen in 17%, with treatment duration being beyond 40 weeks.83
A larger phase II basket trial, KEYNOTE-158, followed the KEYNOTE-028 trial.83 This trial enrolled 104 patients with biliary tract cancers who had progressed or were intolerant to standard therapy. They received pembrolizumab 200 mg intravenously every 3 weeks. A partial response was seen in 6/104 enrolled patients and 17 patients had stable disease. In 31 patients with combined positive score <1, median OS was 7.2 months (95% CI 5.3–11.0 months) versus 9.6 months (95% CI 5.4–12.8 months) in patients with combined positive score ≥1.83
Furthermore, a phase II trial enrolled 54 patients with biliary tract cancer who had progressed after least one line of therapy. Patients were treated with nivolumab until disease progression or unacceptable toxicity.84 The response demonstrated that 10 patients (22%) achieved partial response (one unconfirmed partial response) and 17 patients (38%) had stable disease, accounting for a DCR of 60%. On central review, ORR was just 11%, implying that checkpoint inhibition only has slight anti-tumour activity in heavily pre-treated PD-L1-positive patients with biliary tract cancer.85
Given the limited responses seen with single-agent immunotherapy, studies that combine systemic chemotherapy and other targeted drugs are on-going.85,86 Recently, a randomized, phase II multi-institutional study investigated the role of nivolumab with gemcitabine and cisplatin (Arm A) or nivolumab with ipilimumab (Arm B) in patients with untreated, advanced, biliary tract cancer.86 PFS rate at 6 months, the primary endpoint, was 70.0% in Arm A and 18.6% in Arm B. The median PFS was 8.8 months (95% CI 6.1–11.3 months) in Arm A and 4.1 months (95% CI 2.4–5.2 months) in Arm B. These results suggest ipilimumab has a similar efficacy to gemcitabine/cisplatin in Arm A, although OS estimates are pending maturity.86
Recently, Oh et al. reported results from phase II study assessing the tolerability, efficacy and biomarkers for durvalumab ± tremelimumab in combination with gemcitabine/cisplatin in chemotherapy-naive patients with biliary tract cancer.87 Patients were first enrolled in the biomarker cohort and received 1 cycle of gemcitabine/cisplatin, followed by gemcitabine/cisplatin with durvalumab and tremelimumab every three weeks until disease progression. Following this, patients were randomized to either the three-drug combination of gemcitabine, cisplatin and durvalumab, or four-drug combination of gemcitabine, cisplatin, durvalumab and tremelimumab, until disease progression. The ORR was 50.0% in the biomarker cohort, 73.4% (95% CI 60.5–86.3%) in the three-drug combination and 73.3% (95% CI 60.4–86.2) in the four-drug arm. The median OS was 15.0 months (95% CI 10.7–19.3 months) in the biomarker cohort, 18.1 months (95% CI 11.3–24.9 months) in the three-drug cohort and 20.7 months (95% CI 13.8–27.6 months) in the four-drug arm. While these results are provocative, it should be noted that these results are in an all-Korean population.87
TOPAZ-1 was a large phase III multinational study in which patients were randomized to one of two arms: durvalumab plus gemcitabine and cisplatin for up to 8 cycles, followed by durvalumab until disease progression (Arm A); or placebo plus gemcitabine and cisplatin for up to 8 cycles, followed by placebo until disease progression (Arm B).83 Patients were stratified by disease status (initially unresectable versus recurrent) and primary tumour location. The primary endpoint was OS. This study has completed accrual and is awaiting final survival data (ClinicalTrials.gov identifier: NCT03875235).88
KEYNOTE-966 is a randomized, double-blind phase III study of pembrolizumab plus gemcitabine and cisplatin versus placebo plus gemcitabine and cisplatin as first-line therapy in patients with advanced and/or unresectable biliary tract carcinoma (ClinicalTrials.gov identifier: NCT04003636).89 Results are awaited.
Combinatorial therapy
Tumour angiogenesis and metastasis is driven by the VEGF pathway. The combination of pembrolizumab and ramucirumab, an VEGF-2 antibody, was evaluated in the KEYNOTE-098 trial with 26 previously treated patients with biliary tract cancer.90 The median PFS in this unselected population was 1.6 months and median OS was 6.1 months. These results imply that in biomarker‐unselected, heavily pre-treated patients, the combination of an VEGF-2 antibody and checkpoint inhibition did not improve survival compared with historical controls.90
Future directions
CCAs are a heterogenous group of cancers that are aggressive and portend poor prognosis. While tumour resection offers the only curative approach, most patients are ineligible due to late stage of presentation. The current treatment algorithm for BTC is based on a back-bone of chemotherapy as first line treatment. However, beyond this, treatment patterns are driven by the presence or lack of molecular mutations. This review described the differences between inter- and intra-tumoural factors, aetiologies, genomic alterations and epigenetic variations that drive these cancers. While we have been able to identify some molecular targets, there remains a large proportion of patients who lack treatment options. As we move forward, we need to design smart clinical trials for this heterogenous population and understand resistance mechanisms with these new targeted therapies.