Apart from skin cancers, prostate cancer is the most common cancer in men living in the US and Europe.1–3 About one in nine men will develop prostate cancer during their lifetime, and, in 2008, 899,000 new cases were diagnosed worldwide.4 Ten to 20 % of all patients will develop castration-resistant prostate cancer (CRPC) within 5 years of follow up.5–7 Approximately 84 % of men with CRPC will present with metastatic disease at time of diagnosis and another 5 % will develop the disease within 2 years after diagnosis of CRPC.8 According to recent analyses, median survival from CRPC diagnosis is 14 months.9
However, new therapeutic options, e.g. abiraterone acetate (AA), enzalutamide (ENZ), cabazitaxel (CBZ) and radium-223, which prolong survival significantly, were not considered in any of these analyses.10–13
Metastatic CRPC (mCRPC) is a characterised by disease progression following surgical or medical castration. The European Association of Urology defines mCRPC as castrate levels of serum testosterone, three consecutive increases in prostate-specific antigen (PSA) resulting in two 50 % increases above the nadir, anti-androgen withdrawal for at least 4 weeks, PSA progression despite secondary hormonal manipulations and/or progression of osseous or soft-tissue lesions.15
Molecular Basis of Castration Resistance
Various molecular mechanisms are involved in the development of CRPC. Amplification of the androgen receptor (AR) was identified as one of the first mechanisms in up to 30 % of the patients after initiation of androgendeprivation therapy (ADT).15 Since then, many other molecular events have been identified as the result of cellular acquisition of mechanisms that might overcome the proliferation-inhibiting effect of serum castrate levels of testosterone. Ligand-independent AR activation, intratumoral androgen synthesis, increased expression of AR messenger RNA (mRNA) and enhanced intracellular conversion of adrenal androgens to testosterone and dihydrotestosterone (DHT) are some mechanisms retaining hormonal sensitivity of prostate cancer cells despite serum castrate levels of testosterone.16–20 Two of the main mechanisms underlying the onset of castration resistance are intratumoral biosynthesis of androgens via 17-hydroxylase-17,20-lyase (CYP17) and androgen-independent activation of the AR, which can be a result of AR overexpression, structural changes of the AR caused by genetic mutations and overexpression of coactivators. 21 CRPC has a heterogeneous morphology, immunophenotype and genotype, demonstrating that ‘metastatic disease’ is a group of diseases between patients and even within the same patient.20–22
Patients with mCRPC have a poor prognosis and those patients with metastases are expected to survive up to 18 to 19 months.14 As patients progress, quality of life deteriorates and, until recently, few treatment options were available. Several new therapies have shown an improvement in overall survival (OS) for patients with mCRPC who have already received chemotherapy with docetaxel.10–13 The impact of these new data on daily clinical practice, treatment sequencing and best care for individual patients is not yet fully understood.
It is the aim of the current article to (1) summarise the data of established treatment options in mCRPC, (2) highlight new developments of medical treatment, (3) provide clinically useful algorithms for daily routine and to (4) point out future developments of medical treatment. For practical reasons, we are going to discuss clinical courses of mCRPC into sections of pre- and post-docetaxel treatment.
mCRPC Current Treatment Options –
Pre-Docetaxel Second-line Hormonal Treatment
Until recently, addition of AR antagonists, such as bicalutamide and flutamide, has been the treatment of choice, as the AR remains active despite castrate serum levels of testosterone achieved during treatment with luteinising hormone-releasing hormone gene (LHRH)-analogues or LHRH-antagonists.15,25 A PSA response, defined as a PSA decrease of at least 50 %, can be expected in up to 30 % of patients with a mean progression-free survival (PFS) of about 6 months. However, until now, no trial has shown a benefit in terms of overall and cancer-specific survival. Currently, the TERRAIN trial is comparing the oncological efficacy of the standard approach of bicalutamide 50 mg/day versus ENZ 160 mg/die in men with progressive mCRPC following first-line androgen deprivation with orchiectomy, LHRH analogues or LHRH antagonists.
Treatment with prednisone or dexamethasone can provide palliative improvements in up to 30 % of the patients. Corticosteroid treatment is also associated with a substantial PSA response of ≥50 % in 20 % to 35 % of men. However, no long-term benefit has been demonstrated with this approach.15,25
Therapy with ketoconazole, which inhibits adrenal and intratumoral 17,20-lyase, 17a-hydroxylase and 11ß-hydroxylase, resulted in PSA responses of 30–50 % and a PFS of 6 to 9 months.25,26 However, due to the new treatment options, such as AA (see below), and the observation that the response to AA is significantly diminished after exposure to ketoconazole and hydrocortisone,15,25 this treatment modality will lose its significance.
Sipuleucel-T
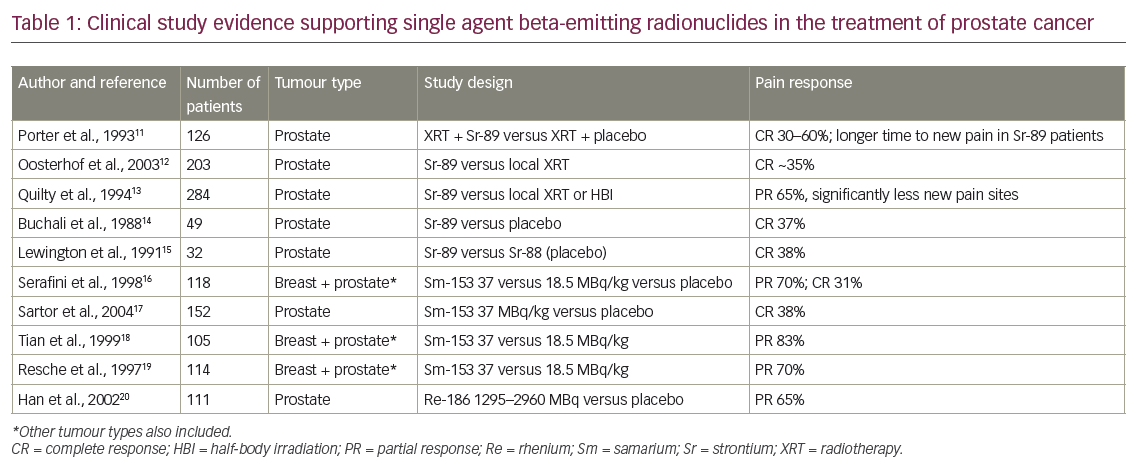
In 2010, sipuleucel-T (Sip-T), an autologous immunotherapy, was approved in the US for treatment of asymptomatic or minimally symptomatic mCRPC patients; however, the drug is not currently available in Europe. Sip-T is an autologous vaccine consisting of individually collected antigen-presenting cells that are exposed to the fusion protein prostatic acid phosphatase and granulocyte colony-stimulating factor (G-CSF) and than reinfused to the patient at week 0, 2 and 4. In the Immunotherapy for Prostate Adenocarcinoma Treatment (IMPACT ) study, median survival with Sip-T was 25.8 months compared with 21.7 months with placebo, and the most frequently reported adverse events were chills, fever and headache in the Sip-T arm.27 It has to be considered, however, that only patients with a good Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, asymptomatic or mildly symptomatic osseous metastases and absence of visceral metastases have been included in the trial (see Table 1).
Abiraterone Acetate
In 2012, AA plus prednisone (AA/P) was approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for treatment of asymptomatic and mildly symptomatic mCRPC patients based on the results of the COUGAR-302 trial.9 AA is an orally administered drug that inhibits 17a-hydroxylase and C17,20-lyase, which are involved in the synthesis of androgens from cholesterol. In the COUGAR-302 trial, 1,088 men with asymptomatic or mildly symptomatic mCRPC were randomised in a 1:1 fashion to either receive AA/P at 1,000 mg/die and 5 mg BID, respectively, or placebo with prednisone. The primary endpoint was overall and radiographic PFS (rPFS) by central review. rPFS was defined as freedom from death from any cause; freedom from progression in soft-tissue lesions as measured with the use of computed tomography (CT) or magnetic resonance imaging (MRI), defined as ‘progressive disease’ according to modified Response Evaluation Criteria in Solid Tumors (RECIST) criteria; or progression on bone scanning according to criteria adapted from the Prostate Cancer Clinical Trials Working Group (PCWG2).28 Secondary endpoints were PSA response, time to opiate use, time to progression, time to chemotherapy and time to deterioration of ECOG performance status. Median OS was 35.3 and 27.2 months in the AA/P and in the placebo group (p=0.01), respectively. Also the co-primary endpoint rPFS was significantly improved in the AA/P with 16.5 months compared with 8.3 months in the placebo arm (p<0.001). On all secondary endpoints AA/P resulted in significantly improved effects. Grade 3 and 4 toxicities developed in 48 % and 42 % of the patients in the AA/P and in the placebo arm, respectively. The most common side effects were fatigue, back pain, arthralgia, nausea and gastrointestinal toxicities. 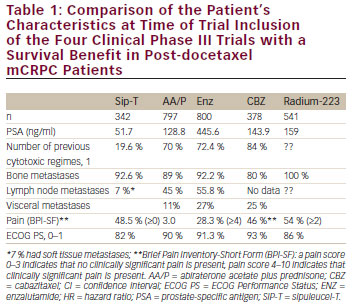
Docetaxel/Prednisone
Docetaxel is a taxane-inducing polymerisation of the microtubules and phosphorylation of the apoptotic oncogene Bcl-2. In 2004 cytotoxic treatment with docetaxel plus prednisone (Doc/P) was the main option for treatment of mCRPC29,30 (see Table 1) once the TA X327 trial demonstrated a significant survival benefit of Doc/P of 3 months compared with mitoxantrone plus prednisone (M/P) in a cohort of 1,006 patients. Since 2010, Doc/P remained the only guideline-recommended first-line therapy with a survival benefit in mCRPC. The median survival was 18.9 months versus 16.4 months (p=0.009) and the 3-year OS rate was 18.6 % versus 13.5 %. It has been shown, quite recently, that Doc/P is active in men with symptomatic mCRPC and especially in patients with poorly differentiated prostate cancer and a Gleason score of 8 to 10. Pain response was also significantly better in the Doc/P arm with a response rate of 35 % compared with 22 % in the M/P arm (p=0.01). Despite the level I evidence and the guideline recommendation, up to 65 % of patients may never receive docetaxel due to poor performance status, comorbidities, concerns about tolerability and, in some cases, patient preference.31,32
None of the combinations with docetaxel have improved oncological outcomes.14 Recently, the results of the Randomized Study Comparing Docetaxel Plus Dasatinib to Docetaxel Plus Placebo in Castration- Resistant Prostate Cancer (READY) trial (docetaxel versus docetaxel plus dasatinib) and the A Multicenter, Randomized, Double Blind Study Comparing the Efficacy and Safety of Aflibercept Versus Placebo Administered Every 3 Weeks in Patients Treated With Docetaxel/ Prednisone for Metastatic Androgen-independent Prostate Cancer (VENICE) trial (docetaxel versus docetaxel plus afilbercept) were disappointing.33,34 The median survival after docetaxel and docetaxel/ dasantinib was 21.2 months versus 21.5 months, respectively, and the median survival after docetaxel versus docetaxel plus afilbercept was 21.1 months versus 22.1 months, respectively.
Post-docetaxel Treatment
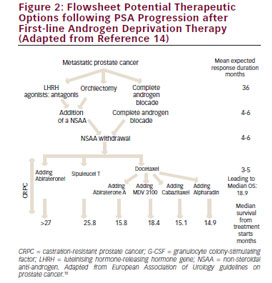
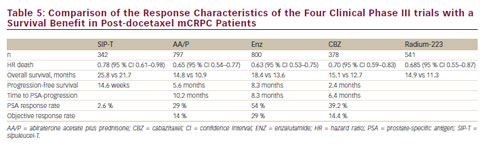
There are several treatment options available for patients who progress following first-line docetaxel chemotherapy for mCRPC: docetaxel rechallenge, AA/P, CBZ plus prednisone, ENZ and radium-223 are active agents in this clinical scenario (see Figure 2and Table 5).
Docetaxel Rechallenge
Although this approach has never been tested in prospective randomised clinical trials, there is enough scientific evidence from large retrospective series to identify patients who may be good candidates for re-exposure.35–38 Patients who respond with a PSA decrease ≥30 % maintained for at least 8 weeks demonstrate a positive PSA response in about 55–60 % during re-exposure without increasing treatmentrelated toxicity.
Abiraterone Acetate plus Prednisone
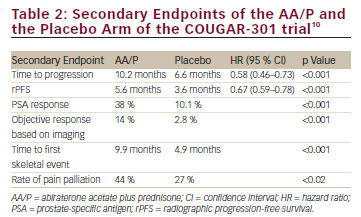
AA/P was evaluated in the clinical phase III, prospective, randomised, double-blind, placebo-controlled multicentre COUGAR-301 trial versus placebo in a cohort of 1,195 progressive mCRPC patients who failed docetaxel-based chemotherapy.11 Patients were randomised in a 2:1 fashion so that 797 and 398 men received AA/P and placebo, respectively. OS was the primary endpoint and time to progression, PSA response and rPFS were secondary endpoints of the trial. The median follow up in the overall study population was 12.8 months. The median treatment duration in the AA/P group and in the placebo group was 8 and 4.5 months, respectively. OS was significantly improved by 3.9 months from 10.9 months in the placebo arm to 14.8 months in the AA/P arm (p<0.001). All secondary endpoints were met and all endpoints demonstrated a significantly improved benefit for the AA/P group (see Table 2).
In terms of treatment-associated toxicity, the profile of adverse events was similar to the COUGAR-302 trial and most of the side effects did not differ statistically significantly between AA/P and placebo. However, adverse events with regard to the CYP 17 blockade (fluid retention, oedema, hypokalaemia, arterial hypertension), cardiac events and elevation of liver transaminases were observed significantly more often in the AA/P arm (55 % vs 43 %; p<0.001). The frequency of fluid retention and oedema (31 % vs 22 %; p=0.04) especially and hypokalaemia (17 % vs 8 %; p<0.001) developed significantly more often in the AA/P arm. The frequency of cardiac events, however, did not differ significantly between the two groups (13 % vs 11 %). Recently, new data have been presented at the two major scientific meetings, namely the Genitourinary-American Society of Clinical Oncology (GU-ASCO) and the annual meeting of the European Association of Urology, which will be discussed below. Goodman et al.39 demonstrated that AA/P is effective even in patients with liver and lung metastases, however, to a lesser degree as in the study cohort. The OS data were 12.9 months versus 8.3 months (p=0.022). Albiges et al.40 described an AA withdrawal syndrome that developed in 32 % of 66 patients who had been treated for a mean period of 5.7 months. Last, but not least, Clayton et al.41 presented data of a populationbased study including 187 mCRPC patients with a mean PSA serum concentration of 138 ng/ml who were with AA/P. The median OS was only 9.3 months and might reflect the oncological efficacy of AA/P in a real-world patient population outside clinical trials.
Enzalutamide (Formerly MDV3100)
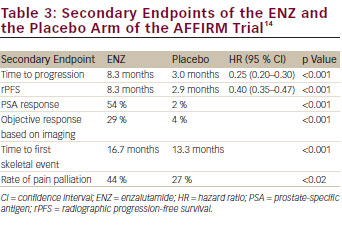
ENZ is an orally administered drug that acts as an AR-signalling inhibitor and is a pure antagonist of the AR with a 10 times greater affinity to the AR relative to bicalutamide.42 ENZ exerts its inhibiting activity at four levels of the AR-signalling pathway: blockade of the AR; inhibiting the nuclear translocation of the AR; inhibiting DNA binding to androgen-response elements; and inhibiting recruitment of coactivators. The AFFIRM trial13 was a clinical phase III, prospective randomised, double-blind, placebo-controlled trial in men with mCRPC, which recruited 1,199 patients who were randomised in a 2:1 fashion to receive ENZ at 160 mg/day or to receive placebo. The primary endpoint of the trial was OS; secondary endpoints were measures of response (PSA decrease, objective remission, quality of life) and measures of progression (time to PSA increase, rPFS, time to first skeletal event). The median follow up was 14.4 months, the median treatment duration was 8.3 months and 3.0 months in the ENZ and the placebo arm, respectively. The median OS was 18.4 months and 13.6 months (p<0.0001) in the ENZ and in the placebo group, respectively, with a relative risk reduction of death by 37 %. All secondary endpoints were met with a statistically significant benefit in the ENZ arm (see Table 3). In terms of safety, the ENZ group experience fewer grade 3/4 toxicities than the placebo group (45 % vs 53 %). There was a higher incidence of all grades of fatigue, diarrhoea, hot flushes, musculoskeletal pain and headache in the ENZ group without any statistically significant differences between the two group. Also, cardiac events (6 % in the ENZ; 8 % in the placebo group), arterial hypertension (6.6 % vs 3.3 %) and liver function abnormalities (<1 %) did not differ significantly between the two groups. The risk of seizures was slightly elevated in the ENZ group with a frequency of 0.6 % vs 0 %. As noted above, data were recently presented at GU-ASCO and the European Association of Urology. Scher et al.43 demonstrated that the use of corticosteroids in parallel to ENZ not only increased grade 3/4 side effects from 34.4 % to 63.3 %, but it also decreased OS to a median time of 11.5 months. These data implicate that ENZ might not be used in patients who need corticosteroids for the management of associated comorbidities and that one of the other second-line therapies, such as AA/P or CBZ/P, might be the drugs of choice. Sternberg et al.44 reported that ENZ is equally effective in elderly people >75 years of age with a median survival time of 18.2 months compared with the placebo group with 13.3 months (p=0.0044). In another presentation, Fleming et al.45 identified clinical parameters associated with long-term response to ENZ. Thirty-five per cent and 22 % of the mCRPC patients in the AFFIRM trial received ENZ for more than 12 months and more than 18 months, respectively. A longer disease history (7.9 vs 5.9 years), a better PSA response (87 % vs 52 %) and a lower metastatic burden were associated with long-term response. These data seem to be important for the decision-making process on the most appropriate therapy on mCRPC patients following docetaxel chemotherapy.
Cabazitaxel plus Prednisone
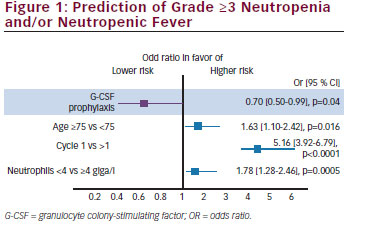
CBZ is a second-generation tubulin-binding taxane that was selected for further clinical trials based on its antitumour activity in models resistant to docetaxel and paclitaxel and its high cytotoxicity. The TROPIC trial was a prospective randomised, open-label clinical phase III trial that recruited 755 patients with mCRPC who progressed during or after docetaxel-based chemotherapy.10 Patients were randomised in a 1:1 fashion to receive CBZ at 25 mg/m2 and prednisone 5 mg BID at 21-day intervals for 10 cycles or mitoxantrone at 12 mg/m2 and prednisone 5 mg BID at 21-day intervals for 10 cycles. The primary study endpoint was OS, secondary endpoints were PFS, PSA-response rate, objective tumour response rate, pain response and safety. The primary endpoint was achieved and chemotherapy with CBZ/P resulted in a median OS of 15.1 months compared with 12.7 months for patients receiving M/P (hazard ratio [HR] 0.70, 95 % confidence interval [CI] 0.59–0.83; p<0.0001). All secondary endpoints of the trials were reached and they were in favour of CBZ (see Table 4). In terms of safety, the CBZ/P group experienced significantly more grade 3/4 toxicities than the placebo group (45 % vs 53 %). The most common side effect were haematological and the most common grade 3/4 toxicities were neutropenia (82 % vs 58 %), leukopenia (68 % vs 42 %) and anaemia (11 % vs 5 %). Diarrhoea was the most common non-hematological side effect and occurred in 6 % and <1 % in the CBZ/P and the M/P group, respectively. This very high frequency of significant treatment-related adverse events could not be reproduced by the German and the European groups when evaluating the safety data of the compassionate-use programmes and the expanded excess programmes.46 The German Compassionate Use Program (CUP) included 111 patients with mCRPC who met the inclusion criteria of the TROPIC trial and the frequency of neutropenia, leukopenia and anaemia decreased to 7.2 %, 9.0 % and 4.5 %, respectively. Grade 3/4 gastrointestinal toxicity was observed in only 0.9 % of the patients. The most likely reason for the improved toxicity profile is the experience of the investigators, guideline-compliant application of G-CSF even at cycle 1 and preventative measures with regard to the treatment of diarrhoea. Heidenreich et al.47 analysed the European CUP including 746 mCRPC patients with regard to the frequency and management of adverse events in senior adults. In that study, 325 (43.5 %) patients were ≥70 years and 145 (19.4 %) men were ≥75 years. The type and the frequency of grade 3/4 side effects did not differ significantly between the younger and the elderly patients except that the frequency of grade 3/4 neutropenia occurred slightly more often in the group of elderly men (19.7 % vs 15 %). Furthermore, G-CSF was used more often at cycle 1 (58.5 % vs 47 %) and throughout CBZ/P treatment (66.8 % vs 58 %) in senior adults. In their analysis, Heidenreich et al.47 developed a risk model to predict grade ≥3 neutropenia and/or neutropenic complications based on a multivariate analysis: age ≥75 years, cycle 1 and neutrophil count <4,000/mm3 before CBZ injection were associated with neutropenic complications (see Figure 1). It has to be noted that even in the presence of these risk factors, prophylactic application of G-CSF significantly reduced the risk of neutropenic complications by 30 % (odds ratio [OR] 0.70, 95 % CI 0.50–0.99; p=0.04). In another study of the French group, Oudard et al.48 demonstrated that CBZ/P is equally effective in poorly differentiated prostate cancer with a Gleason score of 8–10 resulting in a survival benefit of 2.5 months versus M/P (15.2 months versus 12.7 months; p<0.001). 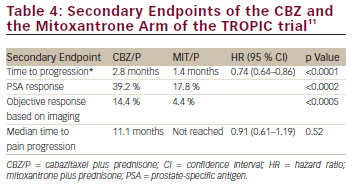
Bone-targeting Agents
More than 90 % of patients with CRPC have bone metastases.29 Bone lesions are associated with elevated osteoclast activity that releases tumour-growth-stimulating factors from the bone. The cycle of bone destruction and tumour growth continues leading to skeletal-related events, such as spinal cord compression, pathological fracture and the need for surgery or external beam radiotherapy.49 Bone metastases are a major cause of death, disability and decreased quality of life, as well as increasing cost of treatment. To date, zoledronic acid is the only bisphosphonate that has been shown to reduce both pain and the number of skeletal-related events in CRPC patients with bone metastases compared with placebo.50,51
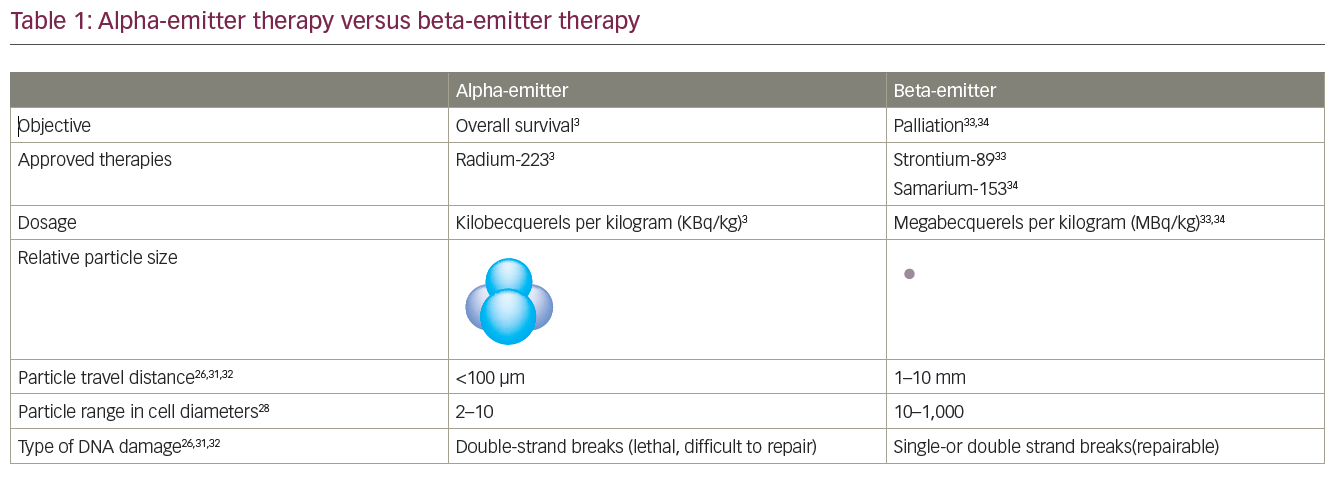
Denosumab is a monoclonal antibody against the receptor activation of nuclear factor kappa-B ligand (RANKL) and is licensed in the US, UK and Europe to treat men at risk of bone loss or fracture associated with hormonal therapy (age >70 years with osteopenia or history of osteoporotic fracture).52 In a recent study of 1,432 mCRPC patients, denosumab significantly increased bone metastasis-free survival by a median of 4.2 months compared with placebo (median 29.5 months vs 25.2, hazard ratio [HR] 0.85, 95 % CI 0.73–0.98; p=0.028). In addition, denosumab also significantly delayed time-to-first bone metastasis (33.2 vs 29.5 months, HR 0.84, 95 % CI 0.71–0.98; p=0.032).54 In a further phase III study, median time-to-first on-study skeletal-related event was 20.7 months with denosumab compared with 17.1 months with zoledronic acid (HR 0.82, 95 % CI 0.71–0.95; p=0.0002 for non-inferiority; p=0.008 for superiority).53 In a recent prospective randomised, double-blind, placebo-controlled trial, Smith et al.54 evaluated the therapeutic efficacy of denosumab 120 mg every 4 weeks versus placebo in 1,423 men with non-metastatic CRPC and aggressive PSA kinetics defined as a PSA >8.0 ng/ml and/or a PSA doubling time <10 months. The primary endpoint of the trial was time-to-first symptomatic or asymptomatic bone metastasis. The median time-to-first bone metastases was significantly prolonged by 4.3 months (29.5 versus 25.2; p=0.028). Bone metastases-free survival was significantly improved by 16 %, 23 % and 29 % in patients with a PSA doubling time of <10, <6 and <4 months, respectively. Rates of adverse events and serious adverse events were similar in both groups, except for osteonecrosis of the jaw and hypocalcaemia. Thirty-three (5 %) patients on denosumab developed osteonecrosis of the jaw versus none on placebo. Hypocalcaemia occurred in 12 (2 %) patients on denosumab and two (<1 %) on placebo. Radium-223 is a radiopharmaceutical that acts as a calcium mimic, and targets new bone growth in and around bone metastases via heavy alpha particles that have an ultra-short range of less than 100 μm. It may take only a single alpha particle to kill a cancer cell, and the short penetration results in highly localised tumour-cell killing with minimal damage to surrounding healthy cells. In the updated analysis of A Double-blind, Randomised, Multiple Dose, Phase III, Multicentre Study of Alpharadin in the Treatment of Patients With Symptomatic Hormone Refractory Prostate Cancer With Skeletal Metastases (ALSYMPCA), which included 921 CRPC patients, the median OS was 14.9 months with radium-223 compared with 11.3 months with placebo (HR 0.695, 95 % CI 0.581–0.8732; p<0.0001).12 Unfortunately, this trial has not yet been published in a peer-reviewed journal.
New and Emerging Developments
Agents Targeting Steroidogenesis
Orteronel (TA K-700) is a lyase inhibitor with selective blockade of 17,20-lyase resulting in fewer mineralocorticoid effects than AA.55,56 In the phase II portion of a dose-finding study, 400 mg BID with prednisone 5 mg BID resulted in a PSA reduction ≥50 % in 52 % of the 96 chemotherapy-naïve recruited mCRPC patients at 12 weeks. The most frequent grade ≥3 were fatigue (76 %) and hypokalaemia (8 %). Due to the selective inhibition of 17,20-lyase over 17a-hydroxylase, 39 patients received orteronel 300 mg BID without corticosteroids in a phase II trial.57 The most common ≥3 adverse events were arterial hypertension (13 %), pneumonitis (18 %), dyspnoea (8 %) and fatigue (5 %) and hypokalaemia (5%). A PSA reduction of 50 % or greater and 90 % or greater was achieved in 45 % and 21 %, respectively. There are two ongoing clinical phase III trials in the pre-chemotherapy and in the post-chemotherapy landscape of mCRPC, which are evaluating the oncological activity of orteronel 400 mg BID in combination with prednisone 5 mg BID versus placebo plus prednisone. In the chemotherapy-naïve trial, a total of 1,454 patients are to be recruited and the primary endpoints for this trial are OS and rPFS. One thousand and eighty-three mCRPC patients are to be recruited in the post-docetaxel trial with OS representing the primary endpoint of the trial. Both trials have completed recruitment.
Galeterone (TO K-001) has combined activity by inhibiting the human CYP17 enzyme, by pure antagonistic activity towards the AR and by inhibiting the binding of androgens to both mutant and wild-type AR.55,58 In the Androgen Receptor Modulation Optimized for Response (AMORI) trial, 49 chemotherapy-naïve mCRPC patients have received different dose regimens from 650 to 2,600 mg for 12 weeks.59 The most common side effects were grade ≤3 and included liver function abnormalities, nausea, diarrhoea and pruritus. A PSA reduction of 30 % or greater and 50 % or greater was achieved by 49 % and 22 %, respectively. Despite the absence of steroid cotreatment no adrenal mineralocorticoid excess was observed. Galeterone has undergone a clinical phase I trial and a phase II trial is on its way.
Androgen-receptor Blocking Agents
ARN-509 is a full antagonist to AR overexpression, which inhibits androgen-dependent gene description and impairs nuclear translocalisation and DNA binding of AR.60 The drug has been tested in a clinical phase I trial recruiting 30 CRPC patients, and a dose of 240 mg daily was identified as the most effective dose.61,62 After 12 weeks of treatment 42 % of the patients had experienced a PSA decline of 50 % or greater. Currently, three prospective randomised clinical phase III trials are on their way including patients with (1) high-risk and non-metastatic CRPC, (2) metastatic treatment-naïve CRPC patients and (3) patients with progression following AA/P. Preliminary results have been presented for parts 1 and 2 and a PSA decline of 50 % or greater was achieved in 91 % and in 88 % of the patients. The most common side effects were tolerable fatigue and gastrointestinal events. ODM-201 is another antiandrogen with similar mechanisms of actions as described for ENZ and ARN-509.63 The potential advantage of ODM-201 is due to that fact that it does not cross the blood–brain barrier and thereby it might prevent the development of seizures. ENZ-4176 is a novel, nucleic acidbased antisense oligonucleotide against AR, which results in selective and specific downregulation of AR mRNA and protein.64
Heat Shock Proteins
Heat shock proteins (HSP) have been identified as AR coactivators and chaperone proteins that are increased after castration in prostate cancer cell lines.65 HSP prevent apoptosis in cancer cells due to a variety of mechanisms and in PCA HSP result in maintaining AR stability, nuclear translocation and transactivation. By binding to the AR, HSP27 facilitates translocation of the AR to the nucleus and binding of the AR to androgenresponsive elements in AR-regulated genes thereby preventing of apoptosis, facilitating metastases and resulting in the development of resistance to chemotherapy. Quite recently, antisense oligonucleotides targeting HSP27 have been developed in vitro and have been evaluated in a clinical phase II trial including 72 patients chemotherapy-naïve mCRPC patients.66 In this prospective randomised trial OGX-427 plus prednisone was evaluated versus prednisone alone. At 12 weeks, 71 % and 40 % of the patients were progression free after OGX-427 or prednisone, respectively. A PSA decline of 50 % or greater was observed in 50 % of the OGX-427-treated patients compared with 20 % in the prednisone group. Furthermore, a measurable disease response occurred in 44 % and 0 % of the OGX-427 and the prednisone group, respectively.
Targeted Therapies
Cabozantinib is another promising bone-targeting agent that inhibits both vascular endothelial growth factor (VEGF) and MNNG HOS transforming gene (MET). MET is upregulated in several tumours and has been shown to drive invasive and aggressive tumours leading to metastases.67,68 MET-driven metastasis may be further stimulated by hypoxic conditions in the tumour environment. In a prospective, randomised, placebocontrolled clinical phase II trial, 171 mCRPC patients were enrolled to receive cabozantinib (100 mg/qd) or placebo. Forty-six of the enrolled patients had previously received chemotherapy whereas the other patients were chemotherapy-naïve.69 Random assignment was halted early based on the observed activity of cabozantinib. Five per cent and 75 % of patients treated with cabozantinib had a confirmed partial response and stable disease, respectively. The median PFS was 29.7 weeks, 23.9 weeks and 5.9 weeks for patients who docetaxel-naïve, docetaxel pretreated and on placebo treatment (p<0.001), respectively. Interestingly, PSA changes did not correlate with the antitumour effects in bone metastases and soft tissue lesions. However, patients with complete (n=14, 12 %) or partial resolution (n=65, 56 %) of bone scans, experienced significantly better response rates to soft tissue metastases compared with men with stable or progressing bone scans (81 % vs 61 %) and they also experienced longer PFS rates at 6 months (56 % vs 48 %, respectively). The most common grade ≥3 side effects were fatigue (16 %), arterial hypertension (12 %), palmar-plantar-erythrodysesthesia (8 %), dehydration (8 %), and pulmonary embolism (7 %). Based on these data, cabozantinib has significant antitumour activity and well tolerable toxicity profile so that it might be well integrated into the therapeutic armentarium to treat mCRPC. Mechanisms of Resistance
As the newly emerging hormonal treatment options are used with increasing frequency in the management of mCRPC patients, it becomes evident that resistance to these drugs develops and that a subgroup of patients never responds to any hormonal manipulation.70–74 Although the exact mechanisms are not completely understood, various hypothesis have been generated based on in vitro and in vivo studies. Primary or acquired resistance to AA/P might be due mutations of the AR facilitating the affinity of the AR for a wider range of other steroid hormones. Some groups have shown that prednisone treatment might activate mutant promiscuous AR and initiate disease resistance.70 The CYP17 enzyme is crucially implicated in the process, which is consistent with the effectiveness of CYP17 inhibitors, such as abiraterone, in CRPC patients.71 Importantly, increased intracellular expression of CYP17 in biopsied metastatic lesions in CRPC patients treated with abiraterone was associated with longer time on treatment and may serve as a tool to predict sensitivity to this class of agents.74 This finding raises the possibility that sustained DHT synthesis might contribute to AA resistance. Resistance to AA/P might also derive from increased intratumoral concentration of DHT and might be overcome by dose escalation of AA/P to inhibit 3ß-hydroxysteroid dehydrogenase.70–74 This approach has been demonstrated to be active in in vitro studies. It also has been suggested that AA/P resistance might be induced by glucocortocoid-induced activation of mutated AR and that iatrogenic steroid mediation, which is mandatory to be added when AA is administered, might induce the resistance. This resistance might be reversed by the addition of pure AR antagonists or by the administration of lyase-inhibitors without the need of steroids such as orteronel or galaterone.
With regard to ENZ, both non-AR mechanisms and AR splice variants have been identified as mediators of resistance and progression.75,76 In xenograft models, increasing concentrations of AR splice variants are associated with disease resistance. Furthermore, loss of the tumour-suppressor gene Pten has been shown to induce resistance to second-generation antiandrogens. Events triggering resistance to ENZ are currently not well established. The increased circulation of AR splice variants might result in such an ENZ escape phenomenon as the increased expression of oestrogen receptor splice variants and the dysregulation of the PTEN/PI3K pathway may play a role. In a recent report comprising 60 patients, cross resistance between AA and ENZ has been demonstrated with a PSA response rate of only 5 % in mCRPC patients who have received AA after progressing on ENZ.
Determining the Sequence of Treatment and Cross Resistance
When selecting second-line therapy, the potential for cross resistance should also be considered. In vitro studies suggest that one mechanism of action for taxanes is inhibition of microtubule-dependent AR translocation to the nucleus, impairing AR signalling and reducing PSA expression.77 Abiraterone impairs AR signalling by reducing CYP17-dependent androgen production, as well as directly binding to AR and reducing AR signalling in a dose-dependent manner. This raises concerns in terms of the potential for cross resistance between microtubule inhibitors and hormonal therapies, such as abiraterone, based on the hypothesis that use of AR signalling inhibitors may impair the ability of taxanes to subsequently inhibit this pathway. In a retrospective analysis of 54 patients with abirateronepretreated CRPC, 35 patients progressed on AA/P and subsequently received docetaxel at 75 mg/m2 every 3 weeks with a median number of six cycles being administered.78 Eight patients never responded to AA/P and were considered primary AA-refractory patients. All eight patients were docetaxel-refractory and did not demonstrate any significant PSA response. PSA declines ≥50 % were observed in 9/35 (26 %) patients, a PSA decrease ≥30 % was observed in 13/35 (37 %) patients. Out of 27 patients who experienced a PSA decline ≥50 % with AA/P, nine had a PSA decrease of 50 % or greater with docetaxel. The median time to PSA progression was only 4.5 months and the median OS was 12.5 months. In the TA X327 trial, the PSA response was 45 %, the median OS rate was 18.9 months and the objective remission rate was 12 %.29 Clearly, there are significant differences between first- and second-line docetaxel, which supports the hypothesis that docetaxel resistance might be triggered by AR overexpression or AR mutation.
When selecting which patient should receive a particular treatment, it may also be important to consider the dynamic biological changes that have been suggested to evolve during the progression of CRPC.79 Androgen dependence in prostate cancer evolves through four states of the AR, as recently outlined by Bluemn and Nelson.80 In the first state (endocrine androgen dependence), the AR is stimulated by testicular testosterone, which allows the disease to be effectively controlled by luteinising hormone-releasing hormone agonists (castration-sensitive prostate cancer). In the second state (intracrine androgen dependence), the AR is activated by androgens of intracellular origin, which are synthesised either de novo or from adrenal precursors. The CYP17 enzyme is crucially implicated in the process, which is consistent with the effectiveness of CYP17 inhibitors, such as abiraterone, in CRPC patients. Importantly, increased intracellular expression of CYP17 in biopsied metastatic lesions in CRPC patients treated with abiraterone was associated with longer time on treatment and may serve as a tool to predict sensitivity to this class of agents.74 Activation of the AR appears to be independent of ligand binding in the third state (ligand independence, AR dependence), due to the expression of an AR splice variant and cross-talk with other signal transduction pathways such as HER2/neu, interleukin 6 and Src kinase. In the fourth state (androgen independence and AR independence), AR signalling is not responsible for neoplastic proliferation, so any AR-directed treatment is likely to be ineffective. This model to better understand the biology in the different phases of CRPC may also be used to help decide the best therapeutic option and treatment combinations for patient.
Biomarkers to Predict Response to Treatment in mCRPC
Biomarkers can be prognostic, predictive or surrogate in nature or can play multiple roles. A prognostic biomarker provides evidence about a patient’s outcome from a disease independent of therapy, whereas predictive biomarkers estimate the likelihood of response/benefit to a specific therapy.81 The majority of biomarkers reported in mCRPC are prognostic rather than predictive82 and although these are helpful, predictive and surrogate biomarkers would be of greater benefit in making treatment decisions. One of the most common markers used in daily clinical practice is monitoring changes in PSA levels. PSA is easy to measure and there is evidence to support its use with cytotoxic therapy; however, it is not a surrogate marker for OS. PSA can rise after the start of therapy in a minority of patients and novel agents may have PSA-independent benefits.69 Furthermore, some subgroups of prostate cancer do not produce PSA. In addition, there is a very small subset of patients with either low PSA or undetectable PSA who may have anaplastic small cell tumours, in some cases this may be in addition to adenocarcinoma and will require a change of treatment (e.g. platinum-based chemotherapy in combination with hormonal therapy). PSA doubling time (PSA DT) is prognostic of OS, and rapid PSA DT may indicate the need for aggressive therapy.83,84 Bone turnover markers (urine N-telopeptide, bone alkaline phosphatase [AP]) reflect tumour–stromal interaction and the prostate cancer microenvironment and they have been linked to survival in several datasets. However, these markers may be normal in patients with visceral/node disease and even in some patients with bone disease.82 Circulating tumour cells (CTCs) can allow for early detection of progression prior to increases in PSA and are tumour specific. CTCs are a strong prognostic marker and may be validated as a surrogate marker in the future;85 however, only 50 % of patients have detectable levels even when using the FDA-approved CellSearch platform.82 Elevated levels of lactate dehydrogenase (LDH) are thought to be reflective of the underlying tumour burden or aggressive phenotype.86 Disease-related anaemia can also indicate a more aggressive form of disease. Anaemia can be easily monitored but may be a consequence of long-term ADT, renal disease, chemotherapy toxicity, chronic disease, iron deficiency from blood loss, bone marrow infiltration or comorbidity. The degree of anaemia correlates with prognosis and was found to be the strongest prognostic factor for docetaxel-related PSA decline, tumour response rate and OS in mCRPC.83,86 Finally, C-reactive protein (CRP) is a marker of systemic inflammation that may also be prognostic of survival in patients previously treated with docetaxel.87 In daily clinical practice, the following markers are often used to identify those patients who may have aggressive disease: short PSA DT, increases in LDH, AP, CRP, carcinoembryonic antigen and high Gleason scores (9/10). The number of visceral metastases may also suggest aggressive disease; however, as patients treated with novel therapies are living longer, more patients may be developing visceral metastases. In addition, a short duration of response to docetaxel (or ADT), rapid development of resistance to docetaxel or rapid clinical progression often seen as an increase in pain or number of bone lesions can all indicate an aggressive phenotype.20,88
Summary
Management of mCRPC has changed dramatically over the last 5 years with four new therapeutic agents – AA, CBZ, ENZ, radium-223 – resulting in a significant improvement of OS (see Table 5). AA/P or Doc/P represents the treatment of choice in mCRPC following first- and second-line ADT. All four agents will become available as second-line therapy following docetaxel chemotherapy. Since mechanisms of action, response rates and treatment associated side effects differ between the various medications, identification of biomarkers or clinical markers predicting best response to deine of the new agent will be of utmost importance for the future. There are many other new agents that are currently being evaluated in clinical phase II/II trials.