




Anaplastic lymphoma kinase (ALK) is a receptor tyrosine kinase (RTK) belonging to the insulin receptor superfamily. The ALK gene is located on the short arm of chromosome 2 and was first identified as an oncogene activated by chromosomal translocation t(2;5)(p23;q35) in anaplastic large cell lymphoma (ALCL) patients.1,2 ALK is normally expressed only in the nervous system. Analysis of in situ hybridization of ALK messenger RNA (mRNA) in mice showed that ALK is predominantly expressed in specific regions of the nervous system, such as the thalamus and midbrain, suggesting that ALK plays an important role in the development and maintenance of the central and peripheral nervous systems.3 Constitutive activation of ALK, derived from chromosomal rearrangements, mutations, or amplification of the ALK gene, has been linked to tumorigenesis and progression of certain cancers such as non-small cell lung carcinoma (NSCLC), breast cancer, and neuroblastoma.4–7 In this article, we discuss the recent advances in the understanding of resistance to ALK, and possible therapeutic targets that overcome this resistance.
Structure of the Anaplastic Lymphoma Kinase Receptor
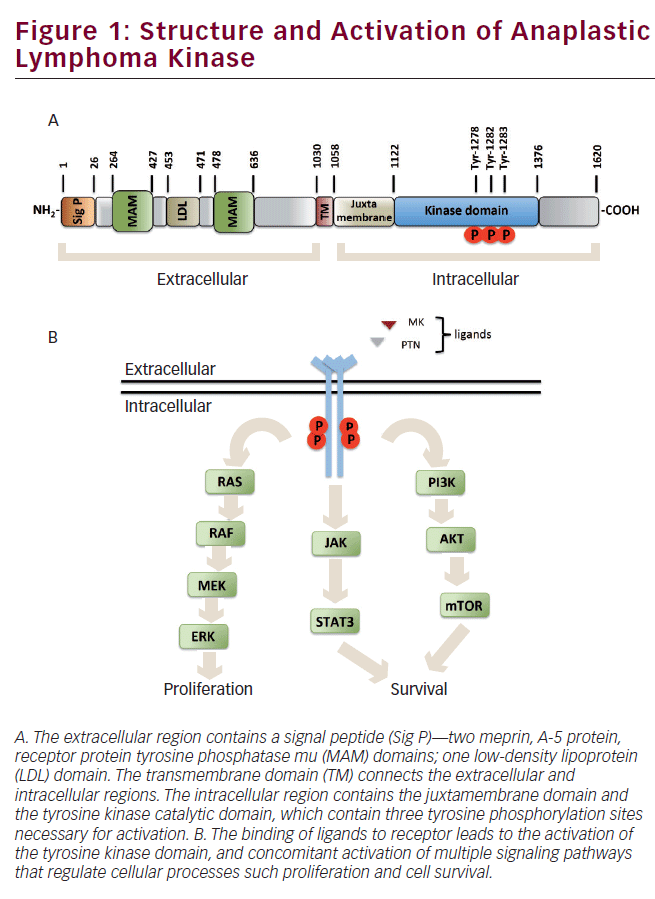
The human ALK gene encodes a 176 kDa protein, which undergoes posttranslational modifications, such as N-glycosylation, altering its migration at approximately 220 kDa on SDS/PAGE.3,8 The ALK receptor is a singlepass transmembrane protein that consists of an extracellular region of 1,030 amino acids (aa), containing an N-terminal signal peptide, two meprin, A-5 protein, receptor protein tyrosine phosphatase mu (MAM) domains separated by a low-density lipoprotein class A (LDL-A) domain, and a glycine-rich region proximal to the transmembrane domain that connects the extracellular region with the intracellular region. The MAM domains of this receptor consist of approximately 160 aa, and are thought to participate in cell–cell interactions,9 whereas the function of the LDL-A domain is still unknown; however, it has been proposed to be involved in ligand recognition.10 The intracellular region contains a juxtamembrane domain and a tyrosine kinase domain. The juxtamembrane domain function in this receptor is still unknown; however, in other receptors, it functions as a modulator of the kinase catalytic activity.11 The kinase domain contains three autophosphorylation sites in tyrosine residues 1278, 1282, and 1283, known as the YXXXYY motif, whose phosphorylation regulates the kinase activity of ALK (see Figure 1A).12,13
Ligands of Anaplastic Lymphoma Kinase
Midkine (MK) and pleiotrophin (PTN) are growth factors considered as putative endogenous ALK ligands capable of acting as autocrine/ paracrine signaling molecules.14,15 MK and PTN are expressed during development of the nervous system and are highly expressed in some cancers where they act as angiogenic factors that drive invasion and metastasis.16,17 MK is a heparin-binding growth factor with a molecular weight of 13 kDa that regulates development of lung, kidney, bone, and nervous systems.18 Stoica and co-workers report that MK stimulates ALK phosphorylation and activates phosphatidylinositol 3-kinase (PI3K) and MAP kinase signal transduction.14 PTN is a 18 kDa protein that acts as a growth factor, regulating neurite outgrowth and proliferation of fibroblasts and endothelial cells.17 Stoica and co-workers report that PTN binds to the extracellular region of ALK, inducing its phosphorylation and activating downstream effectors, such as PI3K (see Figure 1B).15
Anaplastic Lymphoma Kinase Signaling
ALK signal transduction is initiated by binding its ligand, which triggers dimerization and transphosphorylation. Following activation by the ligand, ALK leads to cellular processes involved in oncogenesis. The ALK receptor induces activation of multiple signal transduction pathways. The main pathways activated by ALK are Ras/ERK, JAK/STAT, and PI3K/AKT, which are involved in proliferation, migration, and cell survival (see Figure 1B).
The RAS/ERK Pathway
The extracellular signal-regulated kinase (ERK) controls fundamental cellular processes such as differentiation, proliferation, and migration. ERK signaling is activated by numerous extracellular signals, such as growth factors.19 Stimulation of RTK, such as ALK,20 promotes the exchange of GDP for GTP in the Ras GTPase, which, in turn, recruits Raf kinase to the plasma membrane for its activation.21 Raf kinase activates MEK by its phosphorylation on two serine residues. Active MEK phosphorylates ERK in threonine and tyrosine residues, phosphorylated ERK translocates to the nucleus to modulate gene expression through phosphorylation of transcription factors (see Figure 1B).22
The JAK/STAT Pathway
Signal transducers and activator of transcription (STAT) are a family of latent cytoplasmic transcription factors activated in most cases by growth factor receptors, such as ALK.23,24 The STATs regulate various processes leading to oncogenesis, including angiogenesis, as well as proliferation and survival by regulating the expression of a variety of genes.25 The Janus protein tyrosine kinases (JAKs) are enzymes that mediate activation of STATs in response to growth factors. Stimulation of growth factor receptors promotes activation of JAK through trans-phosphorylation mechanisms. Once activated, JAK phosphorylates to STATs in conserved tyrosine residues, resulting in their dimerization and translocation to the nucleus, where STAT dimers modulate expression of their target genes involved in proliferation and survival (see Figure 1B).26
The PI3K/AKT Pathway
PI3K is a lipid kinase that is activated by RTK such as ALK.27,28 PI3K generates phosphatidylinositol-3,4,5-trisphosphate second messenger that recruits proteins with the pleckstrin homology domain (PH domain) to the plasma membrane. AKT is a serine/threonine kinase with PH domain that plays a vital role in multiple cellular processes, such as proliferation, migration, and survival.29 AKT is activated by phosphorylation on two sites, threonine 308 and serine 473, by the phosphoinositide-dependent kinase (PDK1) and the hypothetical PDK2 kinase, respectively.30 Activated AKT can phosphorylate numerous downstream substrates involved in proliferation and survival (see Figure 1B).31
Anaplastic Lymphoma Kinase as Oncogene
Since the identification of ALK as an oncogene in ALCL, aberrant signaling of ALK in several types of cancer has been reported. This aberrant signaling is associated to chromosomal rearrangements, mutations, or amplification of the ALK gene, leading to a constitutive activation of ALK and phosphorylation of downstream effectors involved in tumorigenesis.
The Role of the Anaplastic Lymphoma Kinase Fusion Protein in Cancer
Chromosomal rearrangements are the most common genetic alterations of the ALK gene that generate fusion proteins. The main features of the ALK fusion proteins are that all retain the intracellular region of ALK, which contains the tyrosine kinase domain, whereas the extracellular region, which contains the ligand-binding site, is replaced by the dimerization domain of the partner protein, the presence of this dimerization domain promotes transphosphorylation of the kinase domains independently of the ligand.32
Lymphoid Cancer
Lymphomas are a type of cancer arising from cells of the immune and lymphatic systems, and they represent 5.3% of all cancers and are the sixth most-common cause of cancer death in the US affecting children and young adults.33
The nucleophosmin-ALK (NPM-ALK) is a fusion protein derived from the t(2;5) (p23;q35) translocation, and approximately 60% of anaplastic largecell lymphoma (ALCL) cases are associated with these rearrangements that generate a constitutively active tyrosine kinase (see Table 1).2,34 Although the molecular mechanisms of NPM-ALK-mediated oncogenesis are incompletely understood, a recent report shows that NPM-ALK promotes survival and cell proliferation through activation of the PI3K-AKT pathway. In this report, it is demonstrated that FOXO3, known as an AKT substrate, regulates survival and proliferation in NPM-ALK-overexpressing cells, where phosphorylation of FOXO3 by AKT promotes their exclusion from the nucleus, preventing the expression of proapoptotic genes and cell cycle inhibitors such as Bim-1 and p27.35 Another type of blood cancer associated with NPM-ALK fusion protein is the diffuse large B-cell lymphoma (DLBCL), where patients diagnosed with ALK-positive DLBCL have a poor prognosis.36,37 Another fusion protein expressed in ALCL is tropomyosin 3 (TPM3-ALK), which is derived from the t(1;2) (q25;p23) translocation (see Table 1). The hybrid protein TPM3-ALK promotes constitutively activation of the kinase domain of ALK, probably through the TPM3 protein–protein interaction domain.38
Lung Cancer
Recently, deregulation of the ALK signaling pathway has been described in many types of nonlymphoid tumors.39 Lung cancers are the most common cause of cancer in the world, and approximately 85% of these tumors are represented by NSCLC.40 In 2007, echinoderm microtubule-associated protein-like 4-ALK (EML4-ALK) was reported as a novel fusion protein found in a subset of patients with NSCLC. EML4-ALK is the result of the inversion on the short-arm chromosome 2 [Inv(2) (p21;p23)] that binds with exons 1–13 of EML4 to exons 20–29 of ALK (see Table 1).4 A recent study indicates that inhibition of the STAT3 and ERK pathways in EML4-ALK-positive cells promotes apoptosis by downregulation of the antiapoptotic protein survivin and upregulation of the proapoptotic protein Bim.41
Breast cancer is a type of cancer arising from the breast tissue and is the primary cause of cancer death in women worldwide. In tumor samples from patients with breast cancer, the presence of EML4-ALK transcripts has been reported in approximately 2% of the total samples; these results were confirmed by fluorescence in situ hybridization (FISH) analysis.5 In support of the hypothesis that aberrant ALK signaling is involved in breast cancer progression, studies show that supernatants of the human breast cancer cell line (MDA-MB-231) contain the putative endogenous ligand of ALK (PTN) and that this ligand stimulates proliferation of endothelial cells.42 These data suggest an important role of wild-type ALK receptor on this disease. In contrast to previous data, a more recent study suggests that EML4-ALK fusion transcripts are not present in breast cancer samples.43
The Role of Wild-type Anaplastic Lymphoma Kinase Receptor in Neural Cancer The neuroblastoma is an embryonal tumor, derived from neural crest cells and is the most common extracranial solid tumor in early childhood.
Many neuroblastomas are incurable with poor prognosis, and these diseases account for 10% of all pediatric cancer deaths.44 ALK is overexpressed in neuroblastoma tumors and neuroblastoma cell lines as a result of gene amplification.45,46 ALK is amplified approximately in 2–3% of neuroblastoma cases.47 In neuroblastoma cells with amplification of the ALK gene, ALK induces hyperphosphorylation of ShcC. The ShcC is a protein that possesses SH2 and PTB domains that function as a scaffolding in activating ERK1/2 and AKT pathways.48,49 In addition, ALK knockdown by small interfering RNA (siRNA) in neuroblastoma cells significantly reduced the phosphorylation of ShcC and AKT, promoting apoptosis.46 These results suggest that amplification of the ALK gene is involved in regulating survival and proliferation in neuroblastoma cells.
The Role of Anaplastic Lymphoma Kinase Mutation in Neural Cancer
In the last decade, activating mutations in the kinase domain of the ALK receptor have been reported. Such mutations induce constitutively the activity of the tyrosine kinase of the receptor, which leads to phosphorylation of downstream effectors, resulting in the regulation of cellular processes such as a proliferation, survival, and migration. It has been reported that ALK is mutated in 8% of all neuroblastoma cases.50 Different germline mutations have been reported in the kinase domain of ALK.6 The R1275 is the most frequent mutation and is detected in 50% of tumors with ALK mutation. In this mutation, arginine (R) is replaced by glutamine (Q) at position 1275, promoting conformational changes in the receptor, breaking the autoinhibitory interaction between the juxtamembrane domain and the kinase domain, and generating a constitutively active receptor.6,51,52 The R1275Q mutant shows higher catalytic activity compared with the wild-type.13 G1128A is another germline mutation, where glycine (G) is replaced by alanine (A) at position 1128, this mutation promotes activation of ERK, AKT, and STAT3, and these mutations are involved in the initiation and progression of neuroblastoma.53 The discovery of activating mutations in germline implicates that these are hereditary; however, mutation penetrance can be incomplete.
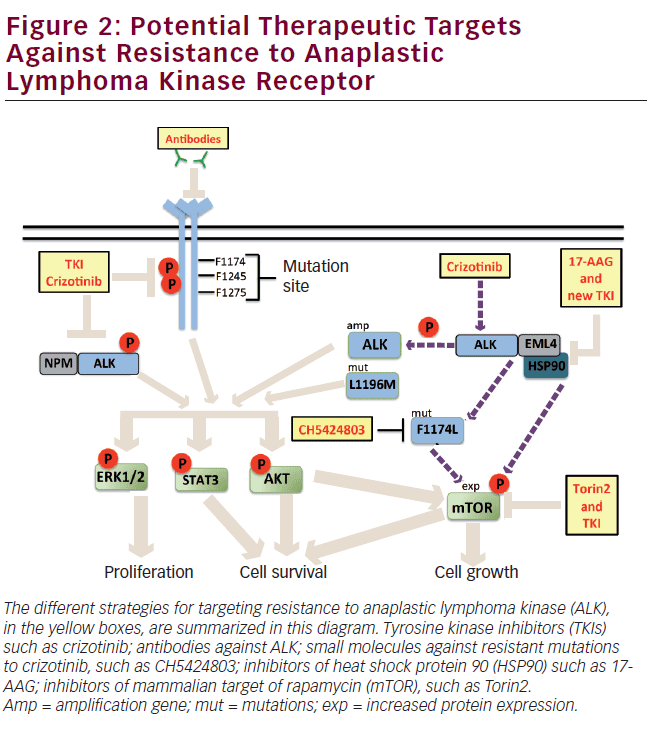
Somatic activating mutations have also been described in the ALK receptor.6 Two mutation hotspots have been found in the kinase domain of ALK: the R1275 and F1174. The R1275Q mutation prevents autoinhibitory interaction.6,52 The F1174L mutation results in the substitution of phenylalanine (F) by leucine (L) at position 1174, this mutation causes resistance to ALK kinase inhibitors.54 Both types of mutations are associated with constitutive phosphorylation of ALK and activation of ERK, AKT, and STAT3 pathways.53,55,56 The third most-frequent mutation in ALK is F1245C, which is also activating (see Table 1 and Figure 2). Treatment with short hairpin RNA against ALK (shRNA-ALK) or small-molecule inhibitor of ALK promotes apoptosis in cell lines that express these mutations; these three mutations represent 86% of all mutations in ALK.13,56,50
Resistance to Anaplastic Lymphoma Kinase Inhibitors
The tyrosine kinase inhibitors (TKIs) could be potential drugs against cancer. Crizotinib is a TKI that inhibits the activity of the tyrosine kinase through competitive binding to the ATP-binding site at the kinase domain. Crizotinib is a drug with high specificity to ALK over other 120 kinases that has been particularly effective against NSCLC, ALCL, and neuroblastoma, which present chromosomal rearrangements or mutation of the ALK gene.57 Currently, crizotinib is being evaluated in phase I clinical trials.54 In patients with EML4-ALK-positive NSCLC, the response rate to crizotinib is high, around 60%.58–60 However, recent reports from EML4-ALK-positive patients have described that these acquire resistance to crizotinib and relapse at 5 months of treatment.61,62 Resistance to crizotinib could be attributed to mutations in ALK, such as mutation F1174L that has been described as resistant to TKI, or to secondary mutations resulting from crizotinib treatment, such as the gatekeeper mutation L1196M, which adopts a structural conformation that decreases binding of the drug to the kinase domain.54 Another mechanism that could lead to resistance is through amplification of the ALK gene (see Figure 2).
Overcoming Anaplastic Lymphoma Kinase Drug Resistance
One of the most common alternatives to overcome resistance to a drug is to increase its concentration in the treatment; however, this strategy is not beneficial in most cases. It has been described that the treatment with increasing doses of crizotinib in EML4-ALK-positive H3122 cell line can result in adverse effects. Treatment with crizotinib at intermediate doses, around 600 nM, has a positive effect on the amplification of ALK, whereas high doses (1 μM) promote mutation F1174L described as resistant to this drug (see Figure 2).61 An alternative to overcome the resistance to crizotinib treatment is the design of drugs based on crystallographic studies of ALK mutant proteins. An example is CH5424802 derived from the benzo[b]carbazole (Chugai Pharmaceuticals Ltd), which inhibits growth of neuroblastoma cells that express the mutant F1174L (see Figure 2).63 Another target strategy for the de novo resistance generated by mutation F1174L is immunotherapy. Recent reports have demonstrated that ALK-directed antibodies inhibit growth of neuroblastoma cells, suggesting their effectiveness in inhibiting the wild receptor and mutants.64 However, the efficiency of these antibodies in tumors with rearrangements of the ALK gene must be questioned, because usually the fusion proteins of ALK show a different cellular distribution from that of the wild receptor,65,66 avoiding antibody binding (see Figure 2). The combination of kinase inhibitors and chaperone inhibitors are another option as targets to overcome drugs resistance. According to the aforementioned, it has been reported that, in tumors expressing the F1174L mutant, the treatment with crizotinib induces differential expression of kinases, decreasing AKT expression, and increasing the expression of mammalian target of rapamycin (mTOR). The combined treatment of crizotinib and Torin2, the latter a selective inhibitor of mTOR, suppresses tumor growth significantly (see Figure 2). In addition, the ALK fusion proteins are proteins known to interact with heat shock protein 90 (hsp90). Inhibition of hsp90 in cell lines that express EML4-ALK induces a significant inhibition of growth (see Figure 2).1 The aforementioned points out that the combined treatment with inhibitors could be the most appropriate strategy to overcome resistance to drugs.
Conclusion
Resistance to drugs, as occurs with the use of TKIs, must inevitably be considered in the research in this field. Elucidation of the resistance mechanisms will allow for the development of new therapeutic strategies to overcome drug resistance in cancer treatment, such as generation of more potent drugs, combination of drugs, or the use of immunotherapy.


