




Acute myeloid leukaemia (AML) is a heterogeneous haematological malignancy characterized by the presence of ≥20% blasts in bone marrow or peripheral blood or the presence of defined genetic abnormalities.1 In 2020, there were an estimated 21,450 new patients with AML and 11,180 AML-related deaths in the USA.2 Most mutated myeloid stem cells proliferate due to genetic alterations, including chromosomal or isolated gene mutations.1 Next-generation sequencing and cytogenetic studies enable risk stratification to inform a therapeutic approach based on the European LeukemiaNet (ELN) 2017 classification (Table 1).3 Nevertheless, the genetic heterogeneity and complexity of AML make risk assessment a challenging task. Overall, AML is associated with poor outcomes, including high relapse rates and a 5-year overall survival rate of 28.3%.4

The backbone of AML treatment consists of induction therapy with cytotoxic drugs or hypomethylating agents (HMAs). For the past 50 years, the standard treatment for AML has included continuous intravenous infusion of cytarabine for 7 days plus an anthracycline (primarily daunorubicin or idarubicin) for the first 3 days; this is called the ‘7+3’ regimen.1 Rates of complete remission (CR) with this regimen range from 55% to 60% in patients younger than 70 years and from 40% to 45% in older patients,5 suggesting that age greatly modulates the response. In addition, the incidence of AML increases with age, so frailty and pre-existing comorbidities represent a challenge to the use of intense induction therapies.6 In elderly individuals, treatment-related mortality (TRM) ranges from 15% to 30%,7 although studies performed in highly specialized cancer centres have reported lower TRM of 3–4%.8 The trend towards decreasing TRM is probably associated with the use of predictive tools, including widespread use of Eastern Cooperative Oncology Group performance status as a proxy for a patient’s ability to tolerate therapy. For those patients who are likely to be unable to tolerate a 7+3 induction regimen, a combination of the HMAs azacitidine or decitabine plus venetoclax is the standard of therapy.9
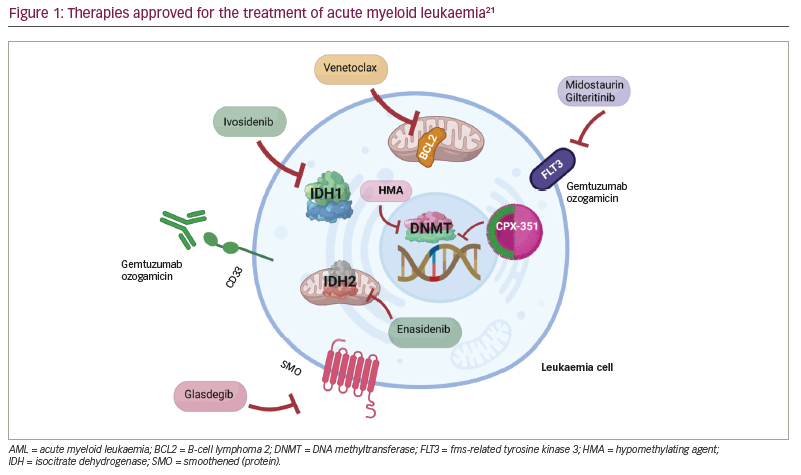
Over the last decade, interest has grown in genetically characterizing AML to provide a more accurate prognosis, personalize management and develop targeted therapies.1,4 The US Food and Drug Administration (FDA) has approved novel therapeutic agents against specific molecular targets, which have improved outcomes (Table 2; Figure 1).10–21 Important benefits of these agents include oral administration and lower toxicity, improved overall response rates (ORRs) (i.e. CR + CR with incomplete haematological recovery [CRi] + partial remission) and a faster treatment response compared with intense chemotherapy. As the widespread use of targeted agents gains momentum, novel combinations with additional targeted or classic AML therapies are likely to evolve.22 In this review, we describe three hypothetical cases of patients with AML to illustrate how an increased understanding of AML genomic alterations has led to therapy developments. In addition, we summarize promising clinical trials that may advance AML management.

Case 1: Treatment of FLT3-mutated acute myeloid leukaemia
A 55-year-old man presented with a petechial rash on his arms, fatigue and splenomegaly. His haemoglobin level was 5.8 g/dL, platelet count 11,000/μL and white blood cell (WBC) count 137,000/μL, with 15% immature myeloid cells. A peripheral blood smear revealed promyelocytes and myeloblasts. The patient’s bone marrow aspirate and biopsy showed 75% cellularity without dysplastic changes. Morphology demonstrated 50% blasts on manual count and 40% blasts by flow cytometry. The blasts were positive for CD34, CD117, CD13 and CD33, consistent with myeloid blasts. Marrow cytogenetics showed a normal karyotype. The gene encoding nucleophosmin 1 (NPM1) was positive for mutations, and fms-related tyrosine kinase 3 (FLT3) internal tandem duplication (ITD) showed a variant allele frequency of 14%.
Treatment option 1: Supportive care
In patients with AML, hyperleukocytosis is defined as a WBC count of >100,000/µL. This complication is associated with adverse outcomes, especially if complicated by leukostasis (i.e. aggregation of leukaemic blasts compromising arterial blood flow). In patients presenting with hyperleukocytosis, induction-associated mortality is about 50% given organ compromise secondary to vascular blast aggregation and ischaemia in the lungs, heart and brain. Leukostasis is a medical emergency requiring cytoreductive therapies such as hydroxyurea, intensive chemotherapy or leukapheresis. Another possible complicating factor during induction therapy is tumour lysis syndrome (TLS), which is caused by an increase in AML cell turnover resulting in electrolyte abnormalities, hyperuricaemia, renal damage and cardiac arrhythmias. TLS prophylaxis involves administering intravenous fluids, allopurinol or intravenous rasburicase. Patients with AML who present with hyperleukocytosis have an increased risk of both TLS and disseminated intravascular coagulation.23
Treatment option 2: Induction chemotherapy
According to the 2017 ELN risk stratification, this patient has intermediate-risk AML, given the presence of mutated NPM1 and FLT3-ITD with a low allele fraction (<0.50).3 NPM1 mutations alone confer a favourable prognosis, but FLT3-ITD correlates with worse outcomes, especially if a high variant allele frequency develops. The combination of both suggests an intermediate-risk profile.3
Given the patient’s young age and lack of comorbid conditions, he received induction therapy with a combination of intravenous idarubicin at 12 mg/m2 daily for 3 days plus intravenous cytarabine at 200 mg/m2 for 7 days (the 7+3 regimen), plus oral midostaurin at 50 mg every 12 hours from day 8 to day 21. After induction, NPM1 reverse transcriptase polymerase chain reaction showed a 2-log reduction in transcript level.
Approximately 60–80% of younger patients treated with 7+3 alone have CR, with a 5-year overall survival (OS) rate of approximately 40%. In comparison, CR is observed in 40–60% of older patients (>65 years) and the OS rate is much lower at about 3% at 5 years.24 While TRM remains a concern, this has declined significantly in recent years given advances in supportive care. Patients presenting with complex cytogenetics, a monosomal karyotype or a mutation in the gene encoding tumour protein p53 (TP53), however, often exhibit poor outcomes with the 7+3 regimen; in these individuals, experimental interventions should be considered.25
Resistance, defined as the absence of CR or relapse from remission, is a common concern in patients with AML. Genetic alterations are the most important predictors of resistance (cytogenetics and mutational status of myeloid-specific genes). Mutations in the genes encoding TP53, additional sex combs-like 1 (ASXL1) and runt-related transcription factor 1 (RUNX1) are adverse predictors. A high allelic ratio (>0.5) for FLT3-ITD is usually considered an adverse predictor according to the ELN 2017 risk stratification. Conversely, mutations in the genes encoding NPM1 and bi-allelic CCAAT enhancer-binding protein alpha (CEBPA) are associated with a good prognosis, unaffected by cytogenetic status.25
FLT3 mutations are seen in approximately 30% of patients with AML, with 25% consisting of ITDs and 5% showing point mutations in the tyrosine kinase domain.26,27 FLT3-ITD mutations are more frequent in patients younger than 60 years than in patients aged ≥60 years (25% versus 15%).25 Both mutations result in constitutive activation of FLT3 signalling via the mitogen-activated protein kinase and phosphoinositide 3-kinase pathways. These abnormalities increase cellular proliferation, decrease apoptosis and block the differentiation of AML cells (Figure 2).10,26 FLT3-ITD AML with an allelic ratio of ≥0.5 is associated with shorter CR duration, increased recurrence and inferior OS.28

Given the targetable nature of FLT3, new therapies have emerged. First-generation FLT3 inhibitors encompass several multikinase inhibitors, including lestaurtinib, sunitinib, sorafenib and midostaurin. Second-generation inhibitors, such as quizartinib, gilteritinib and crenolanib, tend to provide more specific FLT3 inhibition.10 Broad mutational coverage with first generation FLT3 inhibitors is recommended for patients with newly diagnosed FLT3-positive disease to avoid clonal selection, mitigate relapse and decrease the risk of resistance.26 Treatment with single-agent second-generation FLT3 inhibitors, such as gilteritinib, can induce CR in 45–55% of patients with relapsed/refractory (R/R) FLT3-mutated AML by targeting specific clonal restricted relapses.29 More recently, FLT3 inhibitors such as sorafenib have been tested in FLT3-negative AML, given the multikinase inhibitor potential of the drug.25
Midostaurin, a multikinase inhibitor originally developed for the treatment of solid tumours, was approved by the FDA in 2017 in combination with standard induction chemotherapy for FLT3-positive AML based on the results of the RATIFY phase III trial (Table 2).18 The trial included patients aged 18–59 years with detectable FLT3-positive AML. Participants were randomized in a 1:1 ratio to receive oral midostaurin 50 mg twice daily versus placebo during standard chemotherapy induction and consolidation cycles. The 4-year OS rate was 51.4% in patients receiving midostaurin versus 44.3% in the placebo group (hazard ratio: 0.78; p=0.009) in both subgroups of FLT3 mutations and FLT3-ITD with high allelic ratio AML. Superior survival was independent of allogeneic haematopoietic stem-cell transplantation (aHSCT) recipient status. The 4-year event-free survival rate was 28.2% in the midostaurin group versus 20.6% in the placebo group (p=0.044), while disease-free survival was 26.7 versus 15.5 months, respectively (p=0.01). The benefits were observed across FLT3 mutation subtypes. In this study, patients with concurrent NPM1 mutations had a 5-year OS rate of approximately 70%, confirming the favourable prognostic impact.30 Potential side effects of midostaurin include gastrointestinal toxicity, cytopenias and rash.31 There is conflicting evidence regarding QTc interval prolongation in patients with pre-existent cardiac conditions. Nonetheless, close monitoring is advised considering the frequent use of other QTc-prolonging medications such as antiemetics and antifungals in patients with AML.28,32,33
Treatment option 3: Consolidation therapy
The phase II German–Austrian AML Study Group 16-10 trial investigated the effect of midostaurin in induction, maintenance and consolidation therapy in adult patients with FLT3-ITD AML. Participants received one or more induction cycles of 7+3 plus midostaurin, followed by consolidation therapy for those with CR or CRi. Consolidation therapy consisted of oral midostaurin at 50 mg twice daily, followed by aHSCT or four cycles of high-dose cytarabine plus midostaurin. The FLT3 inhibitor was initiated on day 6 and suspended 48 hours before conditioning therapy for aHSCT or subsequent consolidation chemotherapy. Finally, patients received midostaurin (50 mg orally twice daily) as maintenance therapy for 1 year. Although the efficacy of this regimen was similar in younger and older patients, with 72.4% of patients achieving CR/CRi, toxicity was higher in elderly patients, especially those with pre-existent cardiac events.34
The patient described in this case had CR after triple therapy with intravenous idarubicin plus cytarabine and midostaurin. He completed one cycle of consolidation therapy with high-dose cytarabine plus midostaurin, followed by aHSCT. At the time of writing, he had remained disease free for 3 years.
Case 2: Management of IDH-mutated acute myeloid leukaemia in an older patient
A 74-year-old man with a history of heart failure with reduced ejection fraction (New York Heart Association class II stage C), rheumatoid arthritis, uncontrolled type 2 diabetes mellitus and major depressive disorder presented with abdominal distention, postprandial fullness and fatigue. Hepatomegaly was associated with lower extremity oedema. His laboratory results showed a WBC count of 15,000 cells/µL with 87% myeloid-shifted differential, including myeloblasts. Bone marrow evaluation was hypercellular, with 45% of blasts immunophenotypically positive for CD13, CD33, CD34 and CD117. Cytogenetic metaphase analysis showed trisomy 8 (+8) in 14 out of 20 metaphases. Next-generation sequencing demonstrated mutations in the genes encoding isocitrate dehydrogenase 1 (IDH1), Kirsten rat sarcoma viral oncogene homologue (KRAS) and DNA methyltransferase 3 alpha (DNMT3A).
Given the patient’s comorbid conditions, he received oral ivosidenib at a dose of 500 mg/day. On day 12 post-induction the patient developed leukocytosis, shortness of breath and hypoxaemia. Intravenous dexamethasone was administered, given a suspicion of differentiation syndrome. The patient’s symptoms subsided and ivosidenib therapy was continued without major additional complications.
Management of complications: Differentiation syndrome
IDH inhibitor therapy is associated with differentiation syndrome in up to 19% of patients. The complication frequently presents within the first 3 weeks of therapy.35 Differentiation syndrome is a life-threatening adverse event caused by the differentiation of myeloid cells, leading to neutrophilia and a systemic inflammatory response characterized by fever, hypotension, dyspnoea, weight gain, acute kidney injury, pulmonary infiltrates and pleuropericardial effusion.36 It was first described in 1992 in patients with acute promyelocytic leukaemia treated with all-trans retinoic acid.37
In 2020, Norsworthy et al. conducted a systematic analysis of differentiation syndrome in patients with R/R AML treated with ivosidenib and enasidenib.35 This review linked a higher leukaemia burden (defined as higher blast counts in the blood of ≥25% for ivosidenib and ≥15% for enasidenib, or bone marrow blast ≥48%) with an increased risk of differentiation syndrome. Dyspnoea associated with pulmonary infiltrates and pleuropericardial effusions were the most frequent features at presentation, while hypotension was uncommon. Although most cases of differentiation syndrome were of moderate severity, the authors identified two deaths.35 Current data suggest that concurrent mutations in the genes encoding ten-eleven translocation (TET) methylcytosine dioxygenase 2 (TET2) and serine and arginine rich splicing factor 2 (SRSF2) are linked to an increased risk of differentiation syndrome in patients treated with ivosidenib and enasidenib, respectively. The occurrence of differentiation syndrome in patients with AML is associated with a lower CR rate and median OS.38 Management of the condition includes administering intravenous dexamethasone at 10 mg twice daily until the symptoms resolve. The use of hydroxyurea to manage hyperleukocytosis is standard practice.39
Induction therapy
According to the ELN 2017 risk stratification, this patient is at intermediate risk. Older patients pose a therapeutic challenge because they are often refractory to therapy and have a high incidence of adverse genomic features, comorbidities and poor performance status. Universally, elderly patients have limited responses or poor tolerability to intense induction therapy.17 Furthermore, this patient’s comorbidities, particularly his systolic dysfunction, disqualify him from receiving intensive 7+3 induction, given the risk of anthracycline-induced systolic dysfunction.10 Prognostic models to predict TRM are available. Factors other than age, such as performance status, creatinine and albumin levels, and blood counts, may help to identify patients with AML who are at higher risk of complications, and accounting for these additional factors may be superior to simply using age to estimate TRM risk. Indeed, as therapy for elderly patients with AML evolves, it is possible that guideline recommendations will change to more accurately identify patients who will benefit from intense versus less intense interventions. Such risk assessment tools may include novel biomarkers plus chronological age.25 In this patient, the presence of IDH mutations suggested the feasibility of IDH-directed therapy.
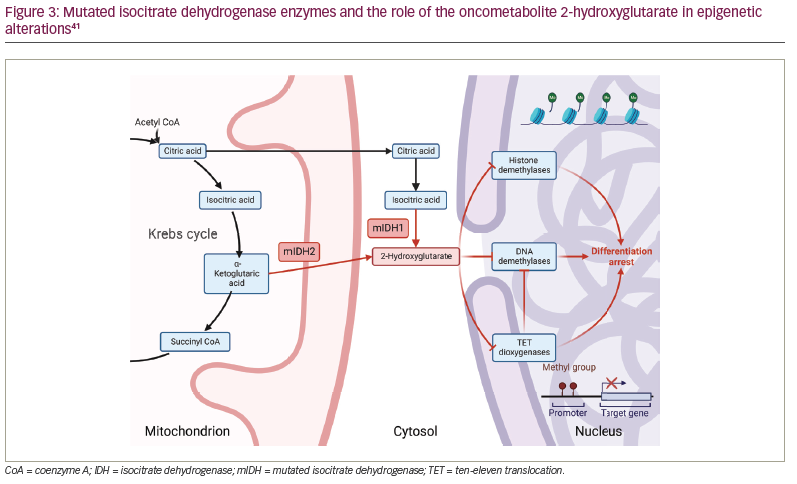
IDH mutations are present in 20% of patients with AML, and around 6–10% involve the IDH1 gene. IDH (IDH1 and IDH2) mutations occur mainly in coding sequences specific to arginine residues within the IDH enzyme. The mutations lead to aberrant enzymatic activity, reducing alpha-ketoglutarate into 2-hydroxyglutarate, an oncometabolite that inhibits the TET dioxygenase family, inducing genome hypermethylation and differentiation arrest (Figure 3).40,41 In addition, IDH mutations correlate with B-cell lymphoma 2 (BCL2) dependency, reinforcing their oncogenic potential. IDH1 and IDH2 are isozymes that share 70% of the same structure but differ in their subcellular location in the cytosol and mitochondrial matrix, respectively.42
Ivosidenib is an IDH1 inhibitor that is approved as monotherapy for patients aged ≥75 years with newly diagnosed AML who are ineligible for standard induction chemotherapy, and for adults with R/R AML (Table 2).40 In a phase I study of patients with newly diagnosed IDH1-mutated AML, ivosidenib resulted in a CR/CRi rate of 42.4%, a CR rate of 30.3% and an ORR of 54.5%.17 Corresponding figures from a phase I study in patients with R/R AML were 30.4%, 21.6% and 41.6%. Median OS was 12.6 months in patients with newly diagnosed disease and 8.8 months in those with R/R disease.43 Enasidenib, an IDH2 inhibitor, has also been studied in patients with newly diagnosed and R/R IDH2-mutated AML, and demonstrated CR/CRi rates of 21% and 27.9%, respectively. CR was achieved by 18% and 18.9% of participants, and the ORR was 30.8% and 39.6%, respectively.17,43,44 Median OS was 11.3 and 8.8 months in newly diagnosed and R/R patients, respectively. Notably, OS differed significantly across response groups, at 22.9 months in those with CR and 10.6 months in those with CRi (p=0.0153).45
Maintenance therapy
Maintenance therapy with ivosidenib should continue until intolerability or disease progression.22 Clinical trials evaluating IDH1 inhibitors as add-on maintenance therapy alongside HMAs in patients with CR after intense chemotherapy or HMA plus venetoclax induction are ongoing (ClinicalTrials.gov identifier: NCT03839771, NCT05010772).46,47
The patient described in this case had CR after three cycles (30 days) of IDH inhibitor. At 12 months of follow-up, he remained transfusion independent and had an absolute neutrophil count of 2,500/μL.
Case 3: Alternative induction for acute myeloid leukaemia
A 72-year-old man with a history of coronary artery disease necessitating bypass presented to the emergency room with fatigue. Physical examination was negative for lymphadenopathy or hepatomegaly. Routine laboratory test showed a WBC count of 20,000/μL, a haemoglobin level of 7 g/dL and a platelet count of 12,000/μL. Bone marrow aspirate and biopsy were consistent with acute leukaemia. Blasts represented 60% of the bone marrow. Flow cytometry revealed CD34, CD117, CD13 and CD33 myeloblasts. The karyotype was normal. Next-generation sequencing showed an NPM1 mutation.
Induction therapy
Elderly patients with AML and significant comorbid conditions who are unable to receive intense induction therapy represent a therapeutic challenge. Patients older than 75 years and those with significant comorbidities are best treated with HMA plus venetoclax.
HMAs are pyrimidine analogues of the nucleoside cytidine that inhibit DNA methyltransferases once incorporated into DNA.48 Azacitidine or decitabine monotherapy is associated with a CR/CRi rate of about 15–30%.49 A response is normally observed after three or four cycles, and patients have median OS of about 10 months.49 The combination of HMA with venetoclax (a BCL2 antiapoptotic protein inhibitor) has demonstrated a composite CR/CRi rate of about 60%.49 Azacitidine plus venetoclax has a median duration of response of 21.2 months and OS of 15.0 months, while the corresponding figures for decitabine plus venetoclax are 16.9 and 16.2 months.9 Most recently, DiNardo et al. administered venetoclax (400, 800 or 1,200 mg/day) with either decitabine or azacitidine to unfit patients with AML (Table 2).19 The combination demonstrated a CR/CRi rate of 66% in the overall population. The CR and CRi rates were, respectively, 41% and 41% in intermediate-risk patients, and 30% and 34% in high-risk patients. The median OS was 17.5 months, with only five of 145 patients dying within 30 days. The trial showed that the 400 mg dose of venetoclax had the best benefit–risk profile.19 A separate study reported comparable outcomes with venetoclax 600 mg plus low-dose cytarabine 20 mg/m2 daily for 10 days, a combination that was approved by the FDA in 2020 (Table 2).25
CR/CRi correlates with mutational and/or cytogenetic status at disease onset, with a more favourable response rate of 91% in patients with NPM1-mutated AML, a lower rate in patients with IDH1/2-positive disease (71%) and the lowest rates in those with poor cytogenetics (60%) and TP53-mutated AML (47%).50 Interestingly, previous data suggest that HMA failure is highly associated with a subsequent inferior 7+3 response.51 HMA therapy results in a similar OS to high-dose cytarabine, with lower toxicity.25 A bone marrow biopsy should be performed after the first cycle of HMA plus venetoclax to evaluate the patient for disease remission.10
The combination of an HMA plus venetoclax is generally well tolerated. Common side effects include gastrointestinal symptoms, fatigue and neutropenia. TLS can also develop, especially in patients who present with a high leukocyte count, elevated lactate dehydrogenase level, hyperuricaemia and underlying kidney disease. Patients should be started at lower venetoclax dose of 20–60 mg (compared with the 100 mg standard starting dose) and with dose escalation following TLS resolution. Hydration and uric acid-lowering drugs (e.g. allopurinol, rasburicase) should be administered to all patients, especially those exhibiting high-risk features such as with hyperleukocytosis.50 Venetoclax can lead to severe myelosuppression. In patients who develop severe neutropenia, supportive therapy with granulocyte colony-stimulating factor plus prophylactic antimicrobial and antiviral therapy is recommended.9 Venetoclax is a substrate of cytochrome P3A4, and up to a 75% dose reduction is needed in patients concomitantly receiving drugs metabolized by the same enzyme, especially if ‘azole’ antifungals are needed as prophylaxis.50
Primary and secondary resistance to HMA plus venetoclax therapy represents an urgent scientific need. Venetoclax resistance is associated with multiple mechanisms, including myeloid leukaemia cell differentiation protein upregulation and stem cells metabolically resetting towards fatty acid dependency rather than oxidative phosphorylation.52 Novel interventions are currently being explored for mitigating the loss of HMA plus venetoclax response.48
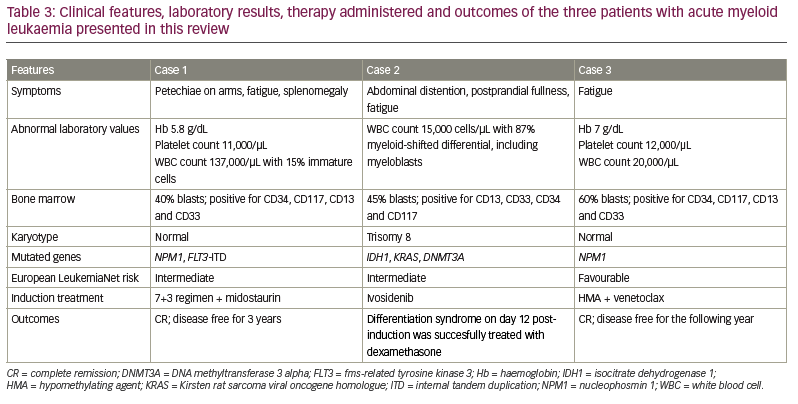
The patient in this case had CR after one cycle of HMA plus venetoclax. The NPM1 mutation became undetectable after two cycles of therapy. He remained transfusion independent and was doing well after 36 months of treatment. A summary of the disease onset, clinical and molecular features, therapies provided and clinical outcomes of the three patients discussed here is given in Table 3.
Alternative maintenance intervention for patients unsuitable for transplantation after intense induction
Oral azacitidine has demonstrated promising results when given as maintenance therapy in patients with AML following intense induction, with or without consolidative high-dose cytarabine. This is an attractive strategy if patients lack a suitable donor or if pre-existing comorbidities restrict their suitability for intense consolidation therapy. The phase III HOVON97 study demonstrated that subcutaneous azacitidine (50 mg/m2 on days 1–5 every 4 weeks until relapse, for a maximum of 12 cycles) significantly increased the 12-month disease-free survival rate compared with controls managed with observation alone (64% versus 42%); however, OS did not differ between the two groups.53 In 2020, the results of the phase III QUAZAR AML-001 study led to FDA approval of oral azacitidine (CC-486) as a maintenance therapy. QUAZAR AML-001 included patients who had experienced CR/CRi after intensive induction and were unable to proceed with intensive curative therapy.54 Compared with placebo, oral azacitidine was associated with significantly longer OS (24.7 versus 14.8 months) and relapse-free survival (10.2 versus 4.8 months). Pharmacokinetic differences between the injectable and oral formulations, including extended drug exposure and sustained orally induced epigenetic modulation, could explain, in part, the lack of an OS benefit in the HOVON97 trial.54
Future directions and goals
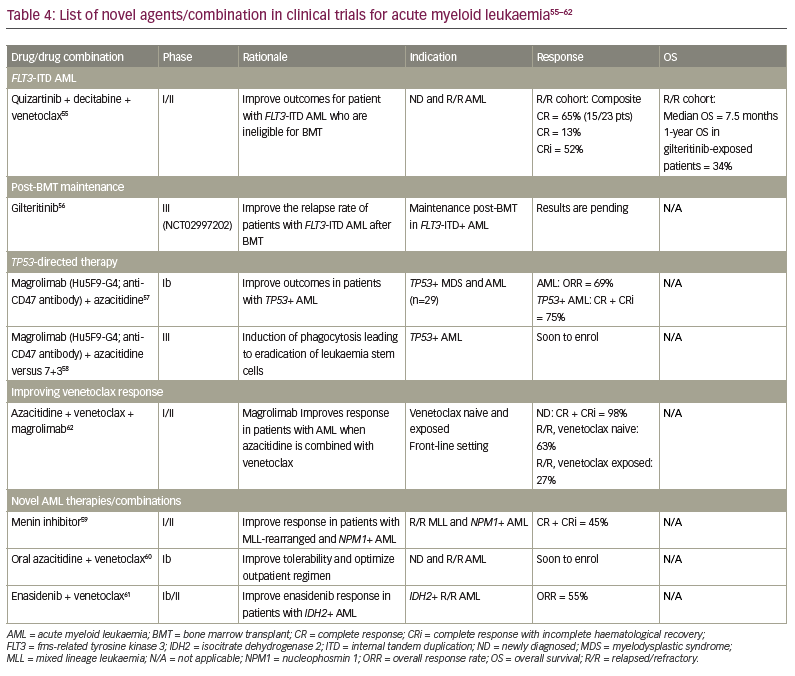
The clinical outcomes for patients with AML have improved over the past 5 years. However, there are still urgent therapeutic needs to consider. First, the results of induction therapy need to be improved for patients with de novo AML in adverse ELN 2017 subgroups, and for those with R/R AML. Second, drug designs that permit oral HMA with improved pharmacokinetics/ pharmacodynamics are required so as to improve leukaemia-free survival in patients without a donor or who are unsuitable for aHSCT. Third, novel drug combinations that can prevent primary and secondary failure of HMA plus BCL2 inhibitor combination therapy are needed. Finally, interventions that improve the relapse risk after bone marrow transplantation, especially in patients in intermediate-risk/adverse ELN subgroups, must be identified. Novel drugs and combinations aimed at addressing some of these needs are outlined in Table 4.55–62
Patients with TP53-mutated AML who are treated with the 7+3 regimen have suboptimal CR rates of 12%. In 2021, liposomal cytarabine plus daunorubicin (CPX formulation) was reported to have a CR rate of 29–32% in patients with AML harbouring TP53 abnormalities (Table 2).11,19 Another study reported a CR rate of 47% with HMA plus venetoclax in the same population.19 However, responses seemed to be short-lived. Magrolimab (Table 4) is a monoclonal antibody designed to block CD47 receptors and induce prophagocytic marrow activity. In a phase Ib trial, magrolimab, in combination with azacitidine, showed an ORR of 69%, with a CR rate of 75% among patients with TP53-mutated AML. Results from phase III trials should determine whether this combination is superior to azacitidine plus venetoclax in inducing CR and reducing measurable residual disease prior to aHSCT.62,63 Recently, Daver et al. presented compelling phase I/II data for the combination of azacitidine, venetoclax and magrolimab in patients with R/R AML.62 The biological basis to using this combination included prolonged survival in vivo of patient-derived xenograft models of AML that were both sensitive and refractory to azacitidine plus venetoclax (Table 4).
Clonal and subclonal relapse are common in AML, even after intense induction/consolidation therapy. This is especially observed among patients in the intermediate-risk and adverse ELN subgroups. The duration of leukaemia-free survival is currently unacceptable among adverse subgroups, highlighting a need for therapies that prevent leukaemia re-emergence. Adaptive metabolic mechanisms may explain, at least in part, primary and secondary resistance to HMA plus venetoclax. Investigators at the MD Anderson Cancer Center and the University of Colorado have proposed that HMA plus venetoclax hinders essential amino acid trafficking in AML cells and effectively eradicates leukaemia-initiated cells that are dependent on oxidative phosphorylation.52 This drug combination is particularly effective in inducing negative measurable residual disease.52 However, resistant AML cells initiate higher fatty acid metabolism dependency after HMA plus venetoclax therapy, suggesting there may be a benefit in exploiting this vulnerability.64 The role of maintenance therapy (oral azacitidine) has been investigated for several years; however, enthusiasm increased after the publication of the QUAZAR AML-001 trial.64 Success in administering oral HMAs has opened a new window of opportunity for AML maintenance therapy. In particular, the ability of patients to reacquire measurable residual disease negativity with extended oral HMA administration (21 days instead of 14 days) was met with enthusiasm by oncologists.64 These observations suggest that HMA dose intensification or administration of an additional antileukaemic agent are promising interventions.
Last but not least, despite impressive results with aHSCT in patients with AML deemed high risk of relapse, the outcomes remain suboptimal for many patients in the ELN 2017 adverse subgroup. Mechanisms resulting in allograft failure and a poor graft-versus-leukaemia effect are not fully understood. Pharmacological interventions to suppress leukemogenesis may synergize with cell therapy to improve patient survival. Attempts to mitigate the proliferative potential of FLT3-ITD clones have paved the development of FLT3 inhibitor trials for maintenance therapy after aHSCT. Indeed, the aHSCT CTN 1506 trial should address gaps in our understanding of the role of FLT3 inhibitor therapy (gilteritinib) following aHSCT.65
We are facing an exciting era of fast-paced drug development in AML. As leukaemia-treating physicians, we should continue to encourage clinical trial participation and foster the integration of basic and clinical science to improve the lives of patients with AML.


