Cancers of the oesophagus and oesophagogastric junction (OGJ), termed upper gastrointestinal (UGI) cancers, are lethal and constitute a significant public health problem. In 2020, 604,000 cases and 544,000 UGI cancer-related deaths were estimated worldwide.1 UGI cancers are the seventh most frequently diagnosed cancer and the sixth most common cause of cancer-related death.1 In the USA, there were 19,260 new cases of UGI cancers and 15,530 UGI cancer-related deaths in 2021, and UGI cancers had the third-lowest 5-year survival rate of any cancer (20%).2
UGI cancers are classified by differences in tumour stage and histology.3 Squamous cell carcinoma is typically located in the upper and middle thirds of the oesophagus and is associated with tobacco and alcohol use.4–7 Adenocarcinoma, whose incidence is increasing in the USA and western Europe, is associated with obesity, gastroesophageal reflux disease and Barrett’s oesophagus.8,9 Treatment paradigms are shifting to include systemic chemotherapy combined with surgery, as well as adopting immunotherapy for all stages.
Tumour-based markers allow investigators to study the direct impact of molecular and immunotherapies on the tumour microenvironment. Efforts to identify biomarkers that predict response to therapy, such as somatic and germline DNA mutations, DNA polymorphisms, radiologic markers, epigenetic alterations and transcriptomic biomarkers, have been limited by small sample size, variability in endpoints and differences in analysis platforms.10–12 Serially measuring biomarkers during treatment is a proposed means to shape trial design with the intent of individualizing therapy.13 Identifying biomarkers that show therapeutic efficacy depends on multiple factors and is difficult in clinical practice due to specific convoluted assay requirements and a lack of generalizable criteria for defining chemosensitivity.14–17
Molecular targets, predictive markers and targeted therapies
The molecular profiling of tumours has led to recent advances in their management. Actionable and predictive markers include but are not limited to human epidermal growth factor receptor 2 (HER2), neurotrophic tyrosine receptor kinase (NTRK) gene fusion, microsatellite instability (MSI) and mismatch repair (MMR), tumour mutational burden (TMB) and programmed death-ligand 1 (PD-L1) expression.18
Human epidermal growth factor receptor 2
HER2 is a receptor tyrosine kinase encoded by Erb-B2 receptor tyrosine kinase 2 (ERBB2) and belongs to the epidermal growth factor (EGFR) family. Overexpression of HER2 leads to increased abnormal cell survival and proliferation via the activation of cell signalling pathways (Ras/Raf/mitogen-activated protein kinase and phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin).19
Immunohistochemistry (IHC) is the initial test for evaluating HER2 expression and measures the membranous reactivity of HER2 on tumour cells.20 HER2 status is graded from 0 (negative or <10% of cells displaying membranous reactivity) to 3+ (positive). Fluorescence in situ hybridization (FISH) is used for equivocal (2+) HER2 IHC status by determining the ratio of copies of the HER2 gene to chromosome 17 centromeres (CEP17) within the nucleus of tumour cells. A HER2:CEP17 ratio of two or greater by FISH is considered positive.20
HER2 expression predicts the benefit of adding trastuzumab (a monoclonal antibody against HER2) to the treatment of patients with OJG (and gastric) adenocarcinoma, which improved survival when combined with first-line chemotherapy in a phase III trial (ToGA study – A study of herceptin (trastuzumab) in combination with chemotherapy compared with chemotherapy alone in patients with HER2-positive advanced gastric cancer; ClinicalTrials.gov identifier: NCT01041404).21 Trastuzumab plus chemotherapy also improved survival compared with chemotherapy alone in the first-line setting for patients with advanced OJG adenocarcinoma in a phase II trial (Efficacy and safety of trastuzumab, capecitabine y oxaliplatine as treatment gastric cancer metastatic [HER2]positive [HerXO]; ClinicalTrials.gov identifier: NCT01503983).22 Another HER2-directed therapy, trastuzumab deruxtecan (an antibody–drug conjugate of trastuzumab with a topoisomerase I inhibitor linked by a tetrapeptide), improved progression-free survival for patients with previously treated advanced or metastatic OGJ or gastric adenocarcinoma in a phase II trial (DS-8201a in human epidermal growth factor receptor 2 [HER2]-expressing gastric cancer [DESTINY-Gastric01]; ClinicalTrials.gov identifier: NCT03329690).23
Notably, an analysis of 295 patients with UGI cancers treated at a single institution who prospectively had next-generation sequencing (NGS) performed on their tumour (and matched blood samples) called into question whether HER2 positivity by IHC/FISH or ERBB2 amplification by NGS better predicted the response to trastuzumab-based therapy.24 In this analysis, 30% of patients with HER2-positive tumours did not have ERBB2 amplification (or had co-mutations in the receptor tyrosine kinase/Ras/phosphatidylinositol 3-kinase pathway) and did not benefit from trastuzumab-based therapy. Patients with higher levels of ERBB2 amplification responded more to trastuzumab-based therapy. Selection for a non-ERBB2 amplified clone in patients who progressed on trastuzumab was theorized to be a mechanism of acquired resistance to trastuzumab-based therapy.24
Neurotrophic tyrosine receptor kinase
NTRK fusions, a genetic target encountered less frequently in UGI cancers, represent another class of therapeutic targets. NTRK2, NTRK2 and NTRK3 genes code for neurotrophic receptors tropomyosin receptor kinase (Trk)A, TrkB and TrkC, respectively. Intra- or interchromosomal rearrangements involving these genes result in the expression of oncoproteins that constitutively activate tyrosine receptor kinases.25 NTRK fusions can be detected using FISH, reverse transcriptase polymerase chain reaction and NGS. To date, completed trials measuring the efficacy and safety of tyrosine receptor kinase inhibitors are limited to those of entrectinib and larotrectinib for treating solid tumours harbouring NTRK gene fusions (including salivary gland tumours, soft-tissue sarcoma, infantile fibrosarcoma, thyroid cancer, non-small cell lung cancer, mammary analogue secretory carcinoma, breast cancer and colorectal cancer). These trials showed clinically meaningful responses, although survival analyses are limited.26,27
Microsatellite instability, mismatch repair, tumour mutational burden and programmed death-ligand 1
Mutations in genes encoding proteins responsible for repairing single base pair insertions and deletions during DNA replication (known as MMR) often occur at short repetitive sequences of DNA known as microsatellites. Such genes include mutL homolog 1 (MLH1), mutS homolog 2 (MSH2), mutS homolog 6 (MSH6) and post-meiotic segregation increased 2 (FMS2). Another mechanism by which MMR is lost – more frequently observed in sporadic tumours than germline tumours – is via promoter hypermethylation of MLH1. Microsatellite mutations, or ‘instability’, in tumour DNA are best assessed by IHC and NGS.
As the cell is unable to repair damaged DNA, mutations tend to accumulate.28 These tumour-specific mutations can be quantified as TMB and are thought to lead to the production of ‘neoantigens’, which the immune system identifies as foreign. This is thought to be the reason why using immune checkpoint inhibitors to disinhibit the immune system leads to a more profound response in tumours with high MSI and deficient MMR.28 Immune checkpoint inhibitors – namely those that block the interaction between PD-L1 (a protein encoded by CD274 found on the surface of many types of cells, including some tumour cells) and programmed death receptor (a protein on the surface of T cells) – disinhibit the T-cell receptor-mediated proliferation of T cells and production of interleukin-2. This leads to pronounced immune-mediated activity against tumour cells that express PD-L1.29 The PD-L1 expression on tumour cells is quantified as a combined positive score (CPS) or the number of PD-L1-positive cells calculated by IHC divided by the total number of tumour cells observed.30 Several phase III trials support the use of immune checkpoint inhibitors in patients with UGI cancers, as outlined in Tables 1–3.31–41
MSI-high (MSI-H)/deficient MMR status generally corresponds with response to immune checkpoint inhibitors in patients with UGI cancers. In NCI-MATCH (Targeted therapy directed by genetic testing in treating patients with advanced refractory solid tumors, lymphomas, or multiple myeloma [the MATCH screening trial); ClinicalTrials.gov identifier: NCT02465060), two of three patients with MSI-H/deficient MMR OJG cancer who received nivolumab achieved an objective response.42 Four of seven patients with MSI-H OJG cancer had an objective response to pembrolizumab in KEYNOTE-059 after progression on at least two chemotherapy regimens (A Study of Pembrolizumab [MK-3475] in participants with recurrent or metastatic gastric or gastroesophageal junction adenocarcinoma; ClinicalTrials.gov identifier: NCT02335411).43 In KEYNOTE-061 (A study of pembrolizumab [MK-3475] versus paclitaxel for participants with advanced gastric/gastroesophageal junction adenocarcinoma that progressed after therapy with platinum and fluoropyrimidine; ClinicalTrials.gov identifier: NCT02370498) and KEYNOTE-062 (Study of pembrolizumab [MK-3475] as first-line monotherapy and combination therapy for treatment of advanced gastric or gastroesophageal junction adenocarcinoma; ClinicalTrials.gov identifier: NCT02494583), patients with MSI-H OJG cancer treated with pembrolizumab had longer median overall survival than those treated with chemotherapy alone in the second-line and first-line settings, respectively.34,35,44
Higher TMB was associated with increased survival after immunotherapy among all cancer types.45 However, when stratified by tumour type, this difference was not observed in patients with oesophagogastric cancers (p=0.164). Nonetheless, a clinical benefit (defined as a radiographic response or stable disease for ≥6 months) was observed among patients with oesophagogastric cancers with a top-quartile TMB treated with immune checkpoint inhibitors compared with patients without a top-quartile TMB.45
PD-L1 expression and CPS are associated with response to immune checkpoint inhibitors. A meta-analysis of 10 randomized controlled trials identified a survival benefit for patients with oesophageal squamous cell carcinoma with CPS ≥10 (hazard ratio 0.60, 95% confidence interval [CI] 0.51–0.70; p<0.01) compared with patients with CPS <10 (hazard ratio 0.83, 95% CI 0.69–1.00; p=0.05).46 Less is known about the predictive nature of PD-L1 expression and CPS in oesophageal and OGJ adenocarcinoma. Tables 1–3 describe large trials that have studied the efficacy of immune checkpoint inhibitors in patients with UGI cancers.31–41
Methods for measuring response to therapy
Imaging
Many assays that assess drug sensitivity have traditionally relied on measuring changes in tumour volume or marker uptake. While studies have evaluated the feasibility and efficacy of measuring residual disease with scintigraphy (e.g. 99mTc-depreotide against somatostatin receptors) and molecular imaging (e.g. nanoparticles and Raman spectroscopy), fluorodeoxyglucose-positron emission tomography/computed tomography at 4–8 weeks after completing treatment is the test of choice for identifying metastasis after treatment with chemoradiation.47–53
Oesophagogastroduodenoscopy
Oesophagogastroduodenoscopy (OGD) enables direct visualization, tissue retrieval, therapeutic resection and ablation. While some advocate for using OGD following definitive chemoradiation to evaluate for residual tumour, others advise caution given its low sensitivity and poor positive predictive value.54,55 If surgery is planned after chemoradiation, OGD is not typically performed since visualization and tissue retrieval can be performed intraoperatively.
Liquid biopsies
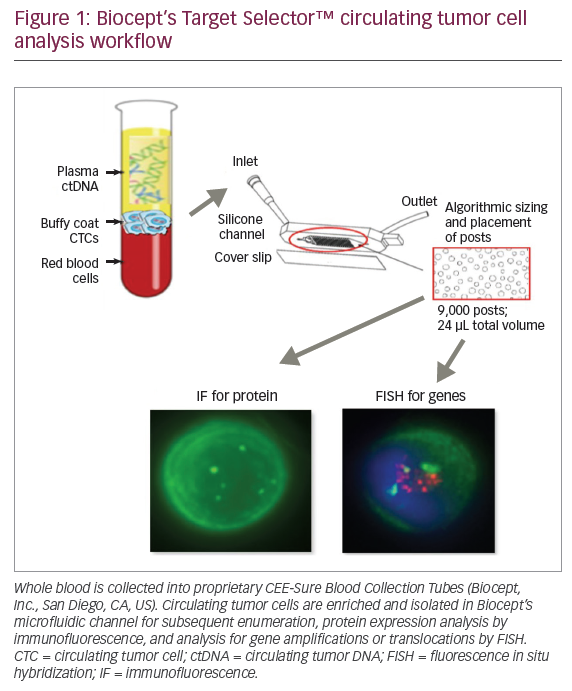
‘Liquid biopsies’ are obtained from peripheral blood, where circulating tumour cells (CTCs) and circulating tumour DNA (ctDNA) are released into circulation via necrosis and apoptosis of solid tumour cells (Figure 1).56 Most commonly, tumour-informed assays, in which NGS is performed on liquid biopsy samples to identify mutational signatures that are similar to the known mutational signature of the primary (or metastatic) tumour, are used. Liquid biopsies have been studied to detect early disease, disease progression, disease recurrence and acquisition of resistance to therapies.57
Compared with ctDNA, CTCs are technically challenging to isolate and sequence, particularly because sampling often yields a low number of cells. However, some suggest that CTCs represent a promising means for discovering novel molecular targets, given that they provide multi-omic information.58 A meta-analysis of eight studies on the presence or absence of CTCs and prognosis in oesophageal cancer concluded that the presence of CTCs is associated with lower overall survival.59 This meta-analysis also identified two studies that evaluated whether the presence of CTCs predicted response to therapy and found that the presence of CTCs was associated with less response to chemoradiation. A second meta-analysis of 18 studies concluded, similarly, that the presence of CTCs was associated with worse progression-free survival, overall survival, and disease recurrence and less response to chemoradiation in oesophageal cancers.60
Cell-free DNA has a high concordance with tumour burden in UGI cancers. In a meta-analysis of 15 studies analysing ctDNA in patients with UGI cancers, the pooled sensitivity and specificity for diagnosing disease were 71.0% (95% CI 55.7–82.6) and 98.6% (95% CI 33.9–99.9), respectively.61 In this same meta-analysis, the pooled sensitivity and specificity for monitoring disease were 48.9% (95% CI 29.4–68.6%) and 95.5% (95% CI 90.6–97.9%), respectively. The most extensive study to date assessing genomic alterations in patients with UGI cancers was a study of 1,630 patients (2,140 tests) with OGJ adenocarcinoma.62 Importantly, the study showed that blood samples obtained from patients with low disease burden yielded lower amounts of ctDNA, that the presence of ctDNA after resection was associated with a higher chance of recurrence and a worse prognosis (in patients with early-stage disease), and that lower baseline quantity of ctDNA and more significant serial decreases in ctDNA after treatment were associated with shorter survival in patients with advanced-stage disease.62 These findings were similar to those of other studies in which post-operative detection of ctDNA was associated with an increased rate of recurrence.63 Additionally, several studies have identified that a high burden of ctDNA detected after chemoradiation correlates with less response to therapy.62,64–67
Identifying changes to tumour biology
Temporal changes to tumour mutational signatures occur in response to systemic therapy. In one analysis of tumour samples (of several cancer types) from 262 patients who underwent genomic testing on two separate occasions, changes in tumour mutational signatures were identified in 66% of patients. Notably, 38% of patients had gained or lost an actionable mutation.68 Temporal heterogeneity in UGI cancers has been identified in several studies. In one analysis of 211 patients with stage II–IV OJG adenocarcinoma, PD-L1 expression and TMB were measured in tumour tissue obtained before first-line chemotherapy and again ≥10 days later. Concordance between pre-treatment and post-treatment PD-L1 expression was 63%, while it was 73% for TMB levels.69
The impact of chemotherapy and radiation on the tumour microenvironment has been studied in several analyses. One study of immunogenomic changes within the tumour microenvironment evaluated 40 patients with oesophageal squamous cell carcinoma (as measured by whole-exome sequencing and whole-transcriptome sequencing), who were treated with 5-fluorouracil, cisplatin and concurrent radiation therapy.70 Post-treatment, patients had increased immune scores (calculated by Estimation of STromal and Immune cells in MAlignant Tumours using Expression) and a decrease in TMB and neoantigen load.70 Other analyses of tissue obtained from patients with gastroesophageal adenocarcinoma after chemoradiation have displayed varying results, ranging from heterogenous tumour-associated immune cell density patterns (i.e. high numbers of CD8+ and PD-1+ cells), a high prevalence of tumoural interferon-γ and upregulation of PD-L1, and acquired mutations in carcinogenic driver mutations.71–74
Intrapatient temporal molecular heterogeneity after treatment resistance was a focus of the PANGEA clinical trial of personalized antibodies for gastroesophageal adenocarcinoma (PANGEA-IMBBP: Personalized antibodies for gastro-esophageal adenocarcinoma – A 1st pilot metastatic trial of biologics beyond progression; ClinicalTrials.gov identifier: NCT02213289).75 In this trial, tumour samples from 27 of 55 patients who progressed on first-line therapy (and had evaluable tumour samples at first progression) had marked intratumoural molecular changes in response to first-line treatment. Similarly, 13 of 27 patients who progressed on second-line therapy (and had evaluable tumour samples at second progression) had further intratumoural molecular changes in response to second-line treatment. Uniquely, this trial customized treatment regimens by adding or substituting monoclonal antibodies and small-molecule inhibitors to target the observed temporal molecular changes, a strategy that led to better 1-year survival and median overall survival compared with historical controls.75
Two other analyses studying the prognostic implications of changes in tumour heterogeneity after chemotherapy in patients with oesophageal adenocarcinoma associated increased intratumour heterogeneity with decreased response to chemotherapy.76,77 An explanation for this may lie in the development of resistance alterations, which was observed in an analysis of 42 patients with gastrointestinal cancers who had cell-free DNA analysis performed after progression on therapy.78 These results suggest that chemotherapy and radiation profoundly affect the tumour microenvironment at the genomic level in patients with UGI cancers, which has implications for future trials incorporating immune checkpoint inhibitors and other novel targeted agents.
A challenge to drawing conclusions from studies investigating the effect of a therapy on the tumour microenvironment is that target inhibition may not correlate with clinical response. Additionally, the impact of treatment often depends on how the drug(s) of interest interacts with components of the signalling pathway and not just the target itself.79,80 This concept was illustrated in a window-of-opportunity study focusing on the pharmacodynamic effects of gefitinib on inhibiting EGFR phosphorylation in tumour samples taken from patients with untreated oesophageal or OJG adenocarcinoma.13 While the pharmacodynamic effects of gefitinib correlated with EGFR phosphorylation, this did not correlate with inhibition of downstream signalling pathways, suggesting that downstream signalling pathways are independent of EGFR activity.13
Conclusions
Opportunities exist for investigators to incorporate serial tissue and liquid biopsies into clinical trials measuring the efficacy of new therapeutic regimens for oesophageal and OGJ cancers. Measuring ctDNA will likely play a more prominent role in prognosticating patients undergoing or completing therapy. More research is needed to determine how changes in tumour biology that occur in response to treatment, as detected by tissue biopsy or liquid biopsy, could further guide the tailoring of therapeutic regimens for patients with advanced oesophageal and OGJ cancers.