
Cancer places a significant and growing burden on healthcare systems around the world; it is the second most common cause of death in the US and accounts for nearly one in four deaths.1 About 1,665,540 new cancer cases are expected in the US in 2014. While population growth and aging will increase the number of new cancer cases in the coming years,2advances in diagnosis will extend the treatment duration required for each patient; the average median duration of treatment with a new drug has risen from 181 days in 1995–1999 to 263 days in 2010–2014.3 Healthcare spending is projected to grow at an average rate of 5.7% during the period 2013–2023, 1.1 percentage points faster than the expected average annual growth in the gross domestic product (GDP).4Cancer care costs are rising faster than overall healthcare costs with cancer drug innovation estimated to reach nearly $100 billion in 2018.5 As a result, the spiraling cost of cancer care, in particular the cost of cancer therapeutics that achieve only marginal benefits, is under increasing scrutiny.6–9
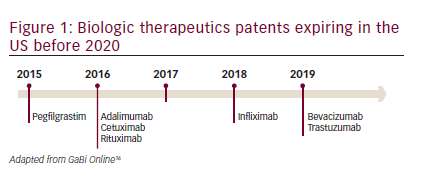
In the past 20 years, recombinant biologics that target specific receptors have made a substantial impact on cancer therapeutics and represent approximately 50% of the pharmaceutical market in oncology.10,11However, these drugs are expensive and are partly responsible for escalating healthcare costs. Year-on-year biologic spending is growing at more than double the rate for small molecule drugs.12In 2010, worldwide sales of all biologics approached the $100 billion mark13and it has been estimated that these could account for more than half of new drug approvals within the next years.14,15However, the patents on biologic therapeutics are beginning to expire (see Figure 1),16allowing the development of biosimilars.17,18Biosimilars represent an opportunity to reduce healthcare spending while increasing access to these important treatments without compromising patient outcomes. Currently, the majority of biosimilar products available for use in oncology are in the supportive care setting; however, their use is increasing rapidly, with an expected shift in focus to agents that offer life-extending benefits, including monoclonal antibodies (mAbs). Biosimilar development programs for therapeutics including bevacizumab, cetuximab, rituximab, and trastuzumab; and supportive care products including epoetin alfa, filgrastim, and pegfilgrastim, are currently ongoing.
This article aims to increase understanding of the biosimilars concept, discuss experience with rigorously evaluated and approved biosimilars in Europe, and to evaluate the status of biosimilars in the US, where the Food and Drug Administration (FDA) has recently implemented a regulatory framework for the evaluation and approval of biosimilars leading to the approval of the first biosimilar in the US.
The concept of biosimilars
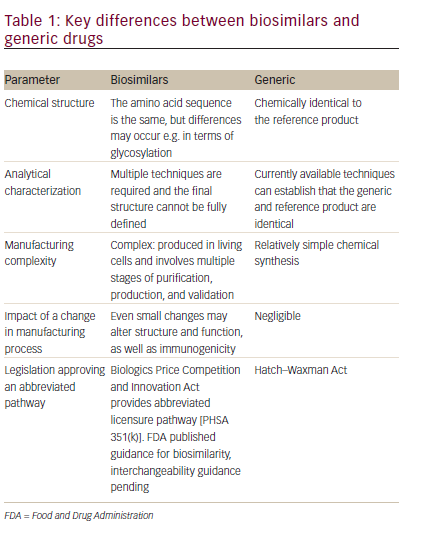
In order to understand the difference between generics and biosimilars, it is important to understand key differences between biologics and small molecule drugs. Biologic therapies are typically large (molecular weight >10,000), complex molecules that, due to their large size, cannot be readily synthesized and are expressed in a living system such as bacteria or, in the case of mAbs, in mammalian cell lines. The manufacturing process is complex and involves multiple stages for cloning; selecting, maintaining, and expanding the cell line; and isolating, purifying, and characterizing the product. The resulting therapies are usually administered by injection or infusion. Small molecule drugs, by contrast, are usually molecules of low molecular weight (<1000), which are synthesized using predictable chemical reactions that can be reproduced and are most frequently administered as oral solids. Therefore, there is inherent variability in the manufacturing of biologics, and while chemical generics can be fully characterized as identical to the originator product, biosimilars cannot (see Table 1)19
An important concept in the manufacture of biologics is variability or post-market changes to a biologic product due to small changes in manufacturing. Biologic therapies are sensitive to manufacturing, storage, and handling conditions and thus have inherent variability, even between batches of the same product. Over the life of a biologic therapy, changes may be introduced to the manufacturing process to improve yield, alter the scale of production or change the source of the individual components. This was demonstrated by analysis of serial batches of biologic agents including darbepoetin, entanercept, and rituximab.20These agents demonstrated minor variations in structure and function. In the example of rituximab, serial batches demonstrated changes in antibody-dependent cell-mediated cytotoxicity (ADCC), which is felt to be a central mechanism in the action of rituximab.21 Variability in biologics is an inevitable consequence of production using biologic systems; these changes are regulated by the International Conference on Harmonisation (ICH) Q5E guidelines that evaluate critical control points in the manufacturing process as well as analytical procedures to evaluate potential differences.22 These guidelines dictate the use of sensitive analytics that are necessary to identify any potentially meaningful differences in the manufacture of a biologic medicine. However, guidelines are not intended to prevent changes over time, rather to ensure that any changes do not alter safety or efficacy. Minor differences have emerged in originator biologic medicine that have been accepted by regulatory agencies. This is a key point to understand in the development of biosimilars for biologic medicines.
Despite the expiration of patents on existing biologics, details of manufacture including bacterial isolates or cell line as well as sourcing of materials remain trade secrets. Furthermore, these methods and materials continue to evolve. Therefore, an attempt by another manufacturer to re-create the originator product will not yield an identical product; the variations in manufacturing can result in differences in molecular structure
(e.g., glycosylation), content (e.g., isoforms, impurities, and aggregates), biologic activity, and immunogenicity. This is the underlying concept of biosimilars: molecules that may have minor differences from the originator product but these are not clinically meaningful.23
The FDA defined biosimilarity as follows: “A biologic product that is highly similar to the reference product notwithstanding minor differences in clinically inactive components … [and] there are no clinically meaningful differences between the biologic product and reference product in terms of safety, purity and potency of the product.”24There are a number of related terms such as ‘me-too biologic’, ‘follow-on biologic’ and ‘biobetters’ but these are not biosimilars and may not have been compared clinically to the reference products. Biosimilars are highly similar to the reference product, other than minor differences to clinically inactive components, and no clinically meaningful differences exist between the biosimilar and originator product in terms of safety, purity, and potency.20Any changes to a biosimilar or a reference product will be constrained by the requirement that the changes do not result in
clinically meaningful differences. Products from different manufacturers will experience change independently, but physical attributes must remain within ranges that result in equivalent safety and efficacy before and after the manufacturing change. These same constraints will likewise limit changes that can occur to interchangeable biologics to only those that do not change safety or efficacy.25
History of biosimilars
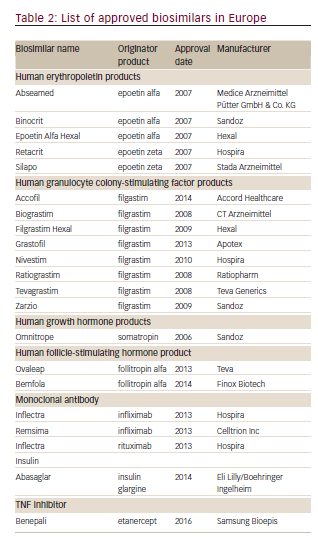
The European Union was the first to develop a separate approval pathway for biosimilars, because of the expiration of patents on several biologics (epoetin alfa, filgrastim, and somatropin) and this has been so successful that it has served as the basis for regulatory guidelines around the world. The European Union established a legal framework for the approval of biosimilars in 2003. In 2005, the European Medicines Agency (EMA) guidelines came into force26 and the first biosimilar, Omnitrope®(Sandoz, Kundl, Austria) was approved in Europe in 2006.27 At the time of writing, a total of 21 biosimilars are approved including the first biosimilar mAb, infliximab (Inflectra®, approved in 2013; Hospira Enterprises B.V., Almere, The Netherlands), with many others currently in development (see Table 2). The companies manufacturing biosimilars are mostly well established biopharmaceutical companies with a track record in the field. The rigorous nature of the process is highlighted by the refusal of marketing authorization for Alpheon®(BioPartners GmbH, Rüsselsheim Germany), a biosimilar recombinant human interferon α-2a. The products displayed different impurity profiles, and clinical trials revealed differences in pharmacokinetics and clinical efficacy compared to the reference product.28
There is now extensive clinical experience with biosimilar epoetin and filgrastim in patients with cancer, and many studies have reported that efficacy is comparable to that of originator products, with no unexpected safety concerns, and substantial economic savings.29–31 A European retrospective review of patients who switched from originator filgrastim to biosimilar filgrastim showed that the latter was effective, clinically comparable to the originator product and prevented dose reductions/ discontinuation in the majority of patients.31
In the US, the Drug Price Competition and Patent Term Restoration Act, also known as the Hatch-Waxman Act, was passed in 1984. This law permitted the manufacture of generic drugs. However, the Hatch-Waxman Act is not applicable to biologics. The FDA has had limited authority to review and approve biosimilars, and this led to delays in the availability of these agents in the US.32The 2010 Affordable Care Act (ACA) enabled the FDA to develop an approval pathway for biosimilars through an abbreviated review process.33 In 2012, draft guidance documents were released, giving clear, stringent guidelines on the permitted variability. A Biologic License Application (BLA) should be submitted to the FDA seeking approval to market any biologic in the US. There are two different types of BLAs: full, stand-alone BLAs filed for approval of an originator biologic product (351(A)),and abbreviated BLAs filed for approval of a biosimilar product (351(K)). In January 2015, the FDA recommended approval of a biosimilar to filgrastim.34Applications for a number of other biosimilars have been filed.
Key stages of biosimilar development and regulation
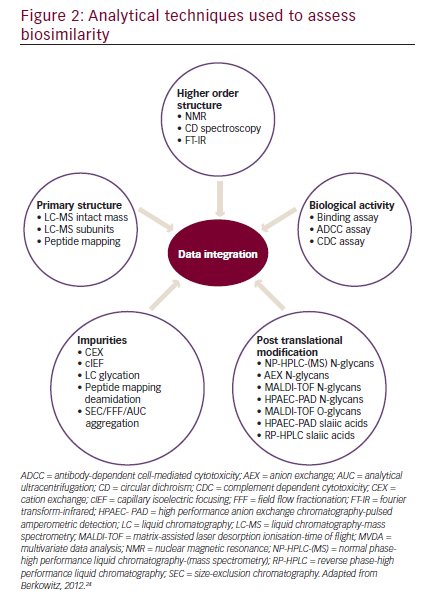
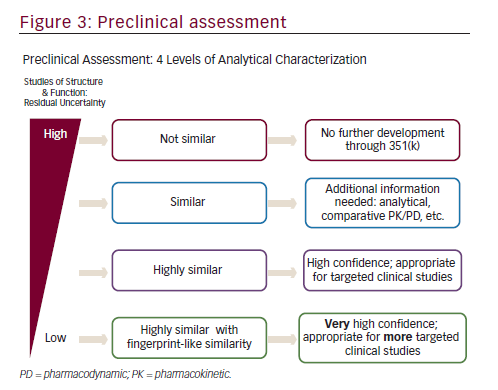
In order to establish biosimilarity, a sponsor must first show analytical data proving that the candidate product is highly similar to the originator product in terms of structure and function.18,24 A biosimilar must have a ‘highly similar’ structure to the originator product in terms of primary structure (identical amino acid sequence is a prerequisite for all classes of biologics, including mAbs), post-translational modifications (PTMs), higher order structure, biologic activity, and purity level (see Figure 2). PTMs of proteins such as glycosylation, oxidation, phosphorylation, sulphation, lipidation, disulphide bond formation, and deamidation may occur during various stages of manufacture such as purification and storage. Even small structural alterations to proteins as a result of PTMs can affect protein activity and may alter the immunogenicity, potentially causing serious safety issues, therefore it is necessary to thoroughly characterize and understand biosimilars during the manufacturing process.24The preclinical analytic package can influence the extent of the clinical data that that will ultimately be needed to gain approval. There are four levels of analytic similarity: not similar; similar; highly similar; or highly similar with fingerprint-like similarity (see Figure 3).24,35Molecules deemed pre-clinically
to be not similar are not candidates for further development in the 351(K) biosimilar pathway. Similar molecules may be required to demonstrate additional pre-clinical analytics as well as a more robust pharmacokinetic (PK) and pharmacodynamic (PD) assessments prior to clinical efficacy evaluation. In contrast, highly similar molecules with fingerprint-like similarity may require only an abbreviated clinical evaluation.
A variety of analytical techniques can provide a quality profile on a mAb with more than 100 attributes, including glycosylation, glycation, and higher order structure (see Figure 2).18,24 In addition, a number of biologic assays allow functional testing methods for mAbs.36An example of physicochemical similarity between the originator rituximab and its biosimilar has been published, including primary and higher order structure, post-translational modifications and size variants, as well as an extensive functional characterization package.37Interestingly, in this analysis the biosimilar GP2013 was analytically more similar to US manufactured Rituxan (rituximab; Genentech, Inc., South San Francisco, CA, US) than Rituxan was to European manufactured MabThera (rituximab; Roche Pharma AG, Grenzach-Wyhlen, Germany). Rituxan and MabThera are both approved as the same agent, rituximab.
Since the originator product varies over its lifetime, multiple analyses should be performed over the shelf life of each batch.18 As discussed
earlier, variations can occur after post-market manufacturing changes. Such variation should be acceptable to regulators and have no impact on the clinical effectiveness or safety of the product.20 A system is in place to ensure that every batch can be tracked. Any parameter for the biosimilar that is outside the known variability of the reference product must be shown to have no impact on the clinical attributes of the final product. Only at this stage can a sponsor conclude that their candidate is ‘highly similar’ to its reference product.
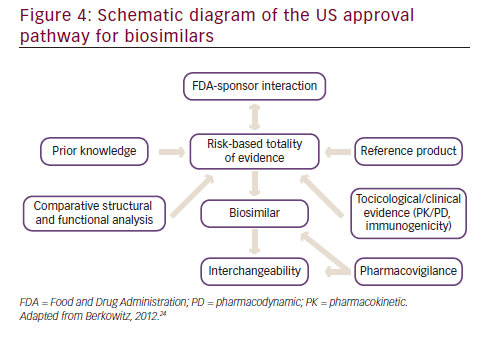
In 2012, the US released a draft document detailing specific steps in obtaining approval for biosimilars (see Figure 4).38 A stepwise approach is taken that involves a substantial level of interaction of the biosimilar sponsor with the FDA. A high level of prior knowledge of the reference product is required. While assessing the comparability of the biosimilar to the reference product, several different lots (of both the biosimilar and the reference product) should be used to assess the variability of the biosimilar and the innovator drug. Structural and functional analysis between the biosimilar and the reference product is required, involving the added use of orthogonal and fingerprint-like methods i.e. state of the art analytical characterization methods that may have not been validated but must be scientifically sound. The more extensive, comprehensive, and robust the comparability process, the lower the likelihood of requiring data from animal and human studies. On the basis of the comparability study data, the level of toxicology and clinical data required is determined.38
Once the pre-clinical evaluation (including comparative animal toxicity if necessary) has demonstrated purity and potency, clinical evaluation can be undertaken.38,39Clinical studies need to include clinical pharmacology studies to demonstrate bioequivalence including assessment of PK or PD (when possible) that are within pre-defined equivalence margins compared to the reference product. Product class-specific PK equivalence will be important to extrapolation decisions that occur later in the development program. Human PD studies should assess measures that are relevant to clinical outcomes; can quickly be assessed with precision and have the sensitivity to detect clinically meaningful differences; and should utilize crossover and parallel designs. An essential aspect of establishing safety is an evaluation of immunogenicity of the biosimilar. This requirement extends into the post-marketing period. Clinical immunogenicity studies evaluate potential differences in the incidence and severity of immune responses using endpoints such as antibody formation (including binding and neutralizing) and cytokine levels.40 Furthermore, a clinical trial must be conducted in an indication approved for the reference product felt to be “most sensitive” to identify differences, if they exist, using an equivalence or non-inferiority trial design.
An important aspect of the 351(K) biosimilar approval pathway is extrapolation. Once a molecule has been approved as a biosimilar on the basis of a single trial in the “most sensitive” indication, it is available for all the approved indications of the reference product but must be scientifically justified and considered on a case-by-case basis. This has raised potential concerns especially when a reference product is used in very different clinical situations. For instance, rituximab is approved for both management of B-cell lymphoma and rheumatoid arthritis (RA). However, the dosing is different in these clinical setting and the extrapolation from RA to lymphoma or lymphoma to RA concerns clinicians.
Following the approval of either a biosimilar or a biosimilar that can be used under the status of interchangeability (see below), a pharmacovigilance program must be initiated to ensure the safety and the effectiveness of the biosimilar product, especially during the initial phase of its public use. The same pharmacovigilance requirements apply to the biosimilar as for the reference product.41Post-approval pharmacovigilance monitoring will include patient registries for assessment of safety issues, voluntary, spontaneous reporting of adverse events (AEs) and medication errors to the manufacturer or FDA by healthcare professionals and patients, as well as mandatory AE reporting by manufacturers to the FDA.41Cost-effective pharmacovigilance programs are evolving, including the Sentinel Initiative holding promise.
Interchangeability
The FDA has also designated a higher standard of biosimilarity, termed interchangeability, requiring data to demonstrate that ‘the biologic product can be expected to produce the same clinical result as the reference product in any given patient and, if the biologic product is administered more than once to an individual, the risk in terms of safety or diminished efficacy of alternating or switching between the use of the biologic product and the reference product is not greater than the risk of using the reference product without such alternation or switch.’38Interchangeability study designs are complex and various designs have been proposed.42Once interchangeability has been established, biosimilars may be substituted at the pharmacy level without the intervention of a healthcare provider.38 However, final FDA guidance on interchangeability is lacking. Individual agents will be evaluated on a case by case basis but in the absence of definitive guidance, designation of a biosimilar as interchangeable is unlikely.
Furthermore, if a biosimilar is ultimately deemed to be interchangeable it does not mean that the pharmacists can freely substitute the biosimilar for the reference product. Rules regarding drug substitution are controlled by the states generally through state pharmacy boards. Legislation has already been passed in 18 states that regulate the use of biosimilars with legislation pending in a number of additional states.43 In most cases, pharmacists are required to notify the ordering physician if a substitution of a prescribed biologic is made with a drug that has approval as interchangeable. In most cases there is also a requirement for patient notification of drug substitution.
Naming
Another area of controversy has been with respect to the naming of biosimilars. Options include: using the same non-proprietary name reference product and any biosimilars; using the same non-proprietary name with a prefix; the same non-proprietary name with a suffix; and using a completely unique non-proprietary names. In the first example of an approved agent in the US, a suffix has been applied to the same non-proprietary name (filgrastim-sndz). However, this is not a final decision. Using the same non-proprietary name communicates the fact that the two products are highly similar. The presence of a prefix may result in failure to identify that the drug was already given by another name, which is also a problem with using a unique non-proprietary name.44,45The white blood cell growth factor Tbo-filgrastim, approved on the 351(A) pathway, using the prefix naming.46The use of a suffix allows for the distinction between reference product and biosimilar and would be grouped together in an electronic health record when sorted by drug name making it clearer what drugs were administered. This issue remains to be resolved.
Clinician acceptance of biosimilars
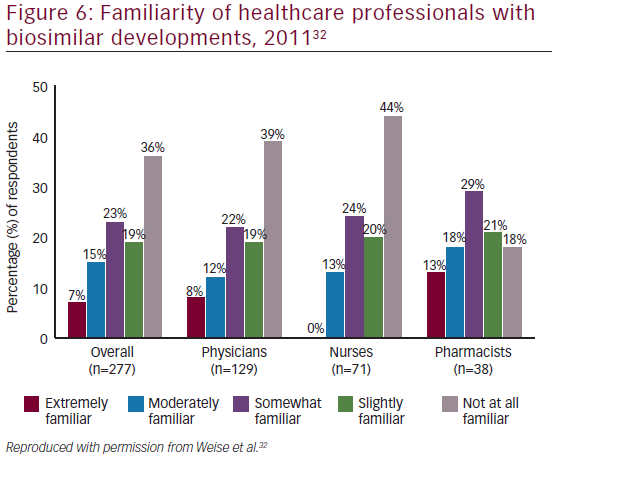
There is considerable interest in biosimilars among the medical community: in a recent survey of physicians in the US, Mexico, Turkey, Russia, and Brazil, almost half of oncologists stated that they would increase the use of human epidermal growth factor receptor 2 (HER2) monoclonal antibody therapy across all treatment settings if a lower cost biosimilar to trastuzumab were available.47However, many misunderstandings also exist regarding biosimilars. At the National Comprehensive Cancer Network (NCCN) 18th Annual Conference in, 2013, a mix of oncology stakeholders (physicians, nurses, pharmacists, and others [n=74]) were surveyed regarding their familiarity of developments surrounding biosimilars, including recent legislation. The majority of respondents (72%) were either ‘not at all familiar’ to ‘slightly familiar’ with developments for biosimilars (see Figure 4).48These findings were similar to those of a 2011 conference survey, in which 277 participants including physicians (n=129), nurses (n=71), pharmacists (n=38), and other types of clinicians or nonpracticing clinicians (n=39) completed a survey. More than half of the respondents were either not at all familiar (36%) or slightly familiar (19%) with recent developments regarding biosimilars (see Figure 5).32
Some of the medical community appear skeptical about biosimilars: initial reports show that physicians are reluctant to switch from the branded infliximab Remicade®(Janssen Biologics B.V., Leiden, The Netherlands) to its biosimilar Inflectra®though there is more willingness to start people on the drug.49Concerns have been expressed among the medical community regarding their efficacy, safety, and impact on reimbursement. One concern is the cost saving they will provide: the cost of developing a biosimilar to reach approval is several orders of magnitude greater than the development of a generic small molecule, largely due to the clinical studies and comparability exercise required to demonstrate biosimilarity. As a result of this investment, cost savings achievable with biosimilars may not be as great as have been experienced with generics.19Concern has been expressed regarding therapeutic indications, for which no specific clinical trials with the biosimilar have been performed and that have been licensed based on extrapolation of efficacy and safety data from other indications.50Strong clinical data will be important for acceptance, and there will be a need for clinician education involving unbiased experts, in the form of national meetings and online education.
Potential benefits of biosimilars
In addition to providing a low-cost alternative to branded products, biosimilars will provide competition that is expected to drive down prices. Although biosimilar development is complex and costly, taking 8–10 years and $100–200 million, biosimilars are likely to cost 20–40% less than their reference products, representing a substantial saving.51,52The greater affordability of biosimilars may result in clinical benefits arising from earlier and wider therapy use, and allow the release of funding for other aspects of cancer care.53It has been estimated that the availability of biosimilars will result in a $44.2 billion reduction in direct spending on biologic agents from 2014–2024, equivalent to 4% of total spending on biologic drugs over the same period.12
Using biosimilars in supportive cancer care could allow additional treatments. A model estimated that the cost savings associated with switching to biosimilar erythropoietin (EPO) in anemia management in EU countries were €110,592,159, translating into an additional 9,770 rituximab, 3,912 bevacizumab, or 3,713 trastuzumab treatments.54,44The increased affordability of biosimilars may also encourage physicians to adhere more closely to clinical guideline recommendations; in a non-interventional study conducted in a community oncology center, switching from originator to biosimilar filgrastim was accompanied by a trend towards increased use of filgrastim as primary prophylaxis.31 In addition, greater uptake of biosimilars may allow greater funding of research to develop improved biologic cancer treatments.
Summary and concluding remarks
In the US we are facing a crisis in health care spending, and although biologic therapeutics do not represent a large percentage of health expenditure, their escalating price is of concern and limit their use. Biosimilar products offer the potential to reduce costs while increasing access to these therapies. Ultimately, the magnitude of the price saving offered by biosimilars is dependent on the final FDA regulations. Oncologists need a full understanding of the regulatory process in order to make informed decisions about incorporating biosimilars into their clinical practice. When evaluating biosimilars, oncologists should consider a manufacturer’s ability to offer a highly similar, safe and efficacious drug product at a cost saving that will encourage healthcare providers to purchase it in preference to the original product to avoid a forced and undesired switching of a patient’s biologic treatment. Increased awareness of these issues is important as biosimilars become incorporated into clinical practice in the US.

