Neuroendocrine neoplasms (NENs) comprise a heterogenous group of malignancies with diverse clinical behaviour determined by tumour characteristics such as grade, differentiation and primary tumour origin.1 While neuroendocrine carcinomas are poorly differentiated aggressive malignancies, well-differentiated neuroendocrine tumours (NETs) tend to be slow growing.2 Despite this, more than 50% of patients with NETs are diagnosed with metastatic disease. Gastroenteropancreatic NETs (GEP NETs) represent the most common subtype of NETs, but the lung is the most common primary site of NETs.3–5
NETs are characterized by overexpression of the mammalian target of rapamycin (mTOR), vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF) pathways, which promote cell survival, proliferation and angiogenesis.1,6 While most NETs are sporadic in nature, they can also arise in association with familial syndromes such as multiple endocrine neoplasia type 1 (MEN1), Von Hippel–Lindau (VHL) syndrome, tuberous sclerosis (TSC) and neurofibromatosis type 1.1 The genetic backdrop of NETs varies based upon primary tumour origin. Pancreatic NETs (pNETs) are typically characterized by inactivation of MEN1, VHL, TSC1 and TSC2 and hyperactivation of the mTOR pathway mediators.6 Small intestinal NETs are typically characterized by mutations in cyclin-dependent kinase (CDK) inhibitor 1 (CDKN1), CDKN2, adenomatous polyposis coli and Kirsten rat sarcoma virus (KRAS), and by copy number alterations of chromosomes 11 and 18.7
The overall survival (OS) for patients with advanced NETs has improved over time, reflecting the increased number of drug licenses and more effective therapeutic options now available.4 Given the increasing disease prevalence and public health burden of NETs, it is important for oncologists to be aware of the advancements in treatment approaches for this growing patient population.4
Peptide receptor radionuclide therapy (PRRT) with lutetium-177-dotatate has become a cornerstone treatment for patients with well-differentiated NETs since its regulatory licensure in 2018;8–10 however, in this review, we will focus on targeted therapies, both in monotherapy and combination settings. Sunitinib and everolimus were the first targeted agents to be approved in the last decade, and since then, many other agents of this class have shown promising clinical activity in patients with NETs.11,12 In this review, we focus on these agents and other drugs that are in earlier phases of development.
Receptor tyrosine kinase inhibitors
NETs are highly vascular tumours, and molecular characterization of NETs identified VEGF as a key oncogenic pathway in these tumours.13 Zhang et al. demonstrated that VEGF overexpression promotes tumour growth by upregulating angiogenesis.13 Additionally, strong VEGF expression was associated with increased metastasis and shortened progression-free survival (PFS) in patients with NENs.13 As such, several receptor tyrosine kinase inhibitors (RTKIs) inhibiting VEGF have been developed for patients with this disease.
Sunitinib
Sunitinib is an oral RTKI that targets VEGF receptors 1–3, FGF receptor 1, PDGF receptors and stem-cell factor receptor, among others.14 The anti-tumour activity of the sunitinib was tested in a two-cohort phase 2 study (A phase II study of the efficacy and safety Of SU011248 in patients with advanced unresectable neuroendocrine tumor; ClinicalTrials.gov identifier: NCT00056693) in patients with extra-pancreatic NETs (epNETs; n=41) and pNETs (n=66).14 Patients were treated with 6-week cycles of oral sunitinib (50 mg/day for 4 weeks, followed by 2 weeks off treatment). While the overall objective response rate (ORR) was 16.7% in patients with pNETs, the ORR in patients with epNETs was 2.4%.
Based on the differential anti-tumour activity demonstrated by sunitinib in pNETs, a randomized, double-blind, placebo-controlled phase 3 trial of sunitinib was launched in patients with this disease (A phase III randomized, double-blind study of sunitinib [SU011248, SUTENT] versus placebo in patients with progressive advanced/metastatic well-differentiated pancreatic Inslet cell tumors; ClinicalTrials.gov identifier: NCT00428597).14,15 A total of 171 patients were randomized to receive either 37.5 mg of daily sunitinib or placebo in addition to best supportive care. Median PFS was 11.4 months in the sunitinib arm and 5.5 months in the placebo arm. The ORR was 9.3% in the sunitinib arm and 0% in the placebo arm. The predominant grade 3/4 adverse events (AEs) were neutropenia (12%), hypertension (10%), hand-foot syndrome (6%), asthenia (5%), diarrhoea (5%) and stomatitis (4%). The results from this study led to sunitinib being licensed in patients with advanced pNETs.16
Cabozantinib
Cabozantinib is another RTKI that targets VEGF receptor 2, mesenchymal epithelial transition, rearranged during transfection (RET), KIT and AXL.17 It was initially tested in a two-cohort phase 2 study in patients with well-differentiated, grade 1–2 advanced pNETs and epNETs (An open-label, phase II study of cabozantinib [XL184] in advanced pancreatic neuroendocrine and carcinoid tumors; ClinicalTrials.gov identifier: NCT01466036).18 Patients with any prior number of therapies were treated at a 60 mg daily dose. The ORR was 15% in both pNET and epNET cohorts. Median PFS was 21.8 months in patients with pNETs and 31.4 months in patients with epNETs. Of the patients who received more than one cycle, 81% required a dose reduction to 40 mg daily. The most common grade 3/4 AEs included hypertension (13%), hypophosphataemia (11%), diarrhoea (10%), amylase or lipase elevations (7%) and fatigue (5%).18 The encouraging PFS and ORR data led to the initiation of the randomized, double-blind phase 3 CABINET (Randomized, double-blinded phase III study of cabozantinib versus placebo in patients with advanced neuroendocrine tumors after progression on prior therapy [CABINET]; ClinicalTrials.gov identifier: NCT03375320) trial.19
The Testing cabozantinib in patients with advanced pancreatic neuroendocrine and carcinoid tumors (CABINET) study is an on-going registration study to assess the efficacy of cabozantinib versus placebo in patients with advanced, well-differentiated pNETs or epNETs who have received at least one previous line of therapy.19 Several amendments have been made to facilitate recruitment for the trial, including removing the need for prior progression on everolimus and allowing for crossover upon progression for patients randomized to the control arm.
Cabozantinib is also being explored in combination with the anti-programmed cell death protein 1 (PD-1) antibody nivolumab in patients with epNETs in a small, single-arm phase 2 study (Phase II trial of cabozantinib in combination with nivolumab for advanced carcinoid tumors; ClinicalTrials.gov identifier: NCT04197310).20
Lenvatinib
Lenvatinib is an oral RTKI targeting VEGF receptors 1–3, FGF receptors 1–4, PDGF receptor α, RET and KIT.21 The anti-tumour activity of lenvatinib was recently reported in the TALENT trial, an open-label phase 2 study (Trial to assess the efficacy of lenvatinib in metastatic neuroendocrine tumors; ClinicalTrials.gov identifier: NCT02678780) that included patients with pNETs (n=55) or epNETs (n=56; all gastrointestinal) who had progressed on at least one prior line of systemic therapy.22 All patients received lenvatinib 24 mg daily until disease progression or treatment intolerance. Patients in the pNET cohort demonstrated an ORR of 42.3%, median PFS of 15.5 months and median OS of 29.3 months. Patients in the epNET cohort demonstrated an ORR of 16.3%, median PFS of 15.4 months, with median OS not reached. The most common grade 3/4 AEs in the pNET cohort were hypertension (21.8%), vomiting (9.1%), abdominal pain (7.3%), diarrhoea (7.3%) and asthenia (5.5%). The most common grade 3/4 AEs in the epNET cohort were hypertension (23.2%), asthenia (16.1%) and diarrhoea (14.3%). Nearly all patients (91.8%) required dose reductions and treatment interruptions.22 Among RTKIs tested in patients with NETs, lenvatinib demonstrated the highest single-agent ORR in the TALENT trial; however, it is unclear whether this was due to differing study populations or more potent inherent anti-tumour activity.
Lenvatinib is being explored in combination with the anti-PD-1 antibody pembrolizumab in patients with epNETs and in a single-arm, phase 2 study (Phase II study of pembrolizumab and lenvatinib in advanced well-differentiated neuroendocrine tumors; ClinicalTrials.gov identifier: NCT03290079).23
Surufatinib
Surufatinib is another oral RTKI and targets VEGF receptors 1–3, FGF receptor 1 and colony-stimulating factor 1 receptor.24 Surufatinib was first tested in patients with well-differentiated grade 1/2 inoperable or metastatic NETs in a phase 1b/2 trial in China (A multi-center, open-label, phase Ib/II clinical trial to evaluate the efficacy, safety, tolerability, and pharmacokinetics of sufatinib in treating advanced neuroendocrine tumors; ClinicalTrials.gov: NCT02267967).24 All patients received surufatinib 300 mg daily until progression or unacceptable toxicity, at which dose interruptions and reductions were allowed. Of the 42 patients with pNETs included in the study, ORR was 19%, disease control rate was 91% and median PFS was 21.2 months. Of the 39 patients with epNETs, ORR was 15%, disease control rate was 92%, and the median PFS was 13.4 months.24
This phase 1b/2 trial was followed by two separate randomized phase 3 trials in China that evaluated the efficacy of surufatinib in patients with pNETs and epNETs. The phase III study of surufatinib in treating advanced extrapancreatic neuroendocrine tumors (SANET-ep) trial (ClinicalTrials.gov identifier: NCT02588170) was a randomized, double-blind, placebo-controlled phase 3 trial in patients with unresectable or metastatic well-differentiated epNETs who had progressed on ≤2 previous lines of therapy.25 Most patients (60.6%) had suspected gastrointestinal primary tumours; 83.8% of these tumours were grade 2. Patients were randomly assigned (2:1) to receive either oral surufatinib 300 mg daily or placebo. Median PFS was 9.2 months in the treatment group versus 3.8 months in the placebo group (hazard ratio [HR]: 0.33; 95% confidence interval [CI]: 0.22–0.50; p<0.0001). The trial was terminated early as it met the predefined criteria for early discontinuation. Treatment-related AEs of grade ≥3 were hypertension (36%), proteinuria (19%) and anaemia (7%).25
Similarly, the Phase 3 study of surufatinib in treating advanced pancreatic neuroendocrine tumors (SANET-p) trial (ClinicalTrials.gov identifier: NCT02589821) was a randomized, double-blinded, placebo-controlled phase 3 trial that included patients with advanced well-differentiated pNETs (86.6% grade 2) who had progressed on ≤2 previous lines of therapy.26 Patients were randomized to receive either surufatinib at 300 mg daily or placebo. The PFS was 10.9 months for the surufatinib group versus 3.7 months for the placebo group (HR: 0.49; 95% CI: 0.32–0.76; p=0.0011). The most common grade 3/4 treatment-related AEs were hypertension (38%), proteinuria (10%) and hypertriglyceridaemia (7%).26
Another phase 1/2 dose-escalation/expansion study conducted in the USA included 32 patients with heavily pre-treated and progressive pNETs and epNETs (A multi-center, open-label, clinical trial to evaluate the safety, tolerability, and pharmacokinetics of surufatinib (HMPL-012), previously named sulfatinib in advanced solid tumors; ClinicalTrials identifier: NCT02549937).27 The maximum tolerated dose was 300 mg daily, consistent with previous studies. An ORR of 9.4% was observed. Sixteen patients (50%) reported grade ≥3 AEs, most commonly hypertension, fatigue, diarrhoea, proteinuria and nausea. This trial established the safety and preliminary anti-tumour activity of this novel agent in patients in the USA. The US Food and Drugs Administration (FDA) has accepted the submission for the new drug application for surufatinib but has yet to license it in the US.
The combined activity of surufatinib with the anti-PD-1 drug tislelizumab is being explored in a multi-cohort phase 2 trial (An open-label phase Ib/II study of surufatinib in combination with tislelizumab in subjects with advanced solid tumors; ClinicalTrials.gov identifier: NCT04579757); two of these cohorts include patients with GEP NETs and lung NETs.28
Pazopanib
Pazopanib targets receptors for VEGF, PDGF α and β, and c-KIT and has demonstrated anti-tumour activity in NETs.29 Pazopanib 800 mg daily was used in a randomized, double-blind, placebo-controlled phase 2 trial (Prospective randomized phase 2 trial of pazopanib [NSC # 737754] versus placebo in patients with progressive carcinoid tumors; ClinicalTrials.gov identifier: NCT01841736) including 171 patients with progressive well-differentiated epNETs (66% with small-bowel NETs).30 Patients in the pazopanib cohort had a median PFS of 11.6 months and median OS of 41 months, while patients treated with placebo had a median PFS of 8.5 months and median OS of 42 months (HR: 0.53; 95% CI: not provided; p=0.0005). The most common grade 3/4 AEs in the treatment arm were hypertension (35%), fatigue (11%), elevations in alanine aminotransferase (10%), elevations in aspartate aminotransferase (10%) and diarrhoea (7%).30
Pazopanib was tested in a phase 1/2 study (A phase I/II study of the combination of temozolomide and pazopanib in advanced pancreatic neuroendocrine tumors [PNET]; ClinicalTrials.gov: NCT01465659) in combination with temozolomide.31 In this study, 28 patients with previously treated advanced pNETs were treated with this combination. Temozolomide was administered on days 1–7 and 15–21, and pazopanib was administered at 400 mg daily. Partial response was observed in 25% of patients, and the disease control rate was 70%. The median PFS was
12.1 months, and the median OS was 36.2 months. Common treatment-related AEs included hepatic dysfunction (16%), nausea (5%) and fatigue (5%). Dose-limiting toxicities included hepatotoxicity and myelosuppression.31
Axitinib
Axitinib is an RTKI that targets VEGF receptors 1–3, PDGF receptor, and c-KIT and has been approved as a first-line treatment of renal cell carcinoma in combination with immunotherapy based on a randomized phase 3 study (A phase III randomized, open-label study to evaluate efficacy and safety of pembrolizumab [MK-3475] in combination with axitinib versus sunitinib monotherapy as a first-line treatment for locally advanced or metastatic renal cell carcinoma [mRCC] [KEYNOTE-426]; ClinicalTrials.gov identifier: NCT02853331).32 Axitinib 5 mg twice daily was tested in a phase 2 trial (A phase II study of axitinib in advanced carcinoid tumors; ClinicalTrials.gov identifier: NCT01435122) in 30 patients with progressive unresectable/metastatic epNETs.33 The median PFS was 26.7 months, while the 12-month PFS rate was 74.5%. The median OS was 45.3 months. The ORR was 3%, and the disease control rate was 73.3%. Grade 3/4 hypertension was the dose-limiting toxicity leading to treatment discontinuation in 6 patients (20%). Although axitinib does seem to possess anti-tumour activity in patients with advanced epNETs, the high rate of dose-limiting hypertension is a potential barrier to its real-world use.33
The AXINET trial (A phase II/III randomized double-blind study of sandostatin LAR in combination with axitinib versus sandostatin LAR with placebo in patients with advanced G1-G2 neuroendocrine tumours [WHO 2010] of non-pancreatic origin; ClinicalTrials.gov: NCT01744249) was a randomized trial that tested octreotide ± axitinib in patients with advanced grade 1/2 epNETs.34 Patients who had received up to 2 lines of prior systemic therapy (except for prior VEGF- or VEGF receptor-targeted drugs) were eligible and were randomized 1:1 to receive octreotide with either axitinib (n=126) or placebo (n=130) until disease progression or unacceptable toxicity. The primary endpoint was PFS. Gastrointestinal tumours were the predominant primary site (40%). Although the ORR was higher in axitinib-treated patients compared with placebo-treated patients (17.5% versus 3.8%, respectively; p=0.0004), there was no statistically significant difference in PFS between the arms (median PFS:17.2 months versus 12.3 months, respectively; p=0.169). Common grade 3/4 AEs included hypertension (21%), diarrhoea (13%), asthenia (9%), cardiac disorders (3.2%) and nausea/vomiting (2%).34
We have summarized the on-going/completed studies of single-agent RTKIs in patients with well-differentiated NETs in Table 1. Studies with RTKIs such as nintedanib and sorafenib are included in this table, though these drugs are not discussed in the preceding paragraphs as these drugs are no longer in development for patients with NETs.35,36 Given the potential synergistic activity of VEGF RTKIs and immunotherapy combinations in other diseases, several trials are exploring this approach in NETs. These trials are also summarized in Table 1.
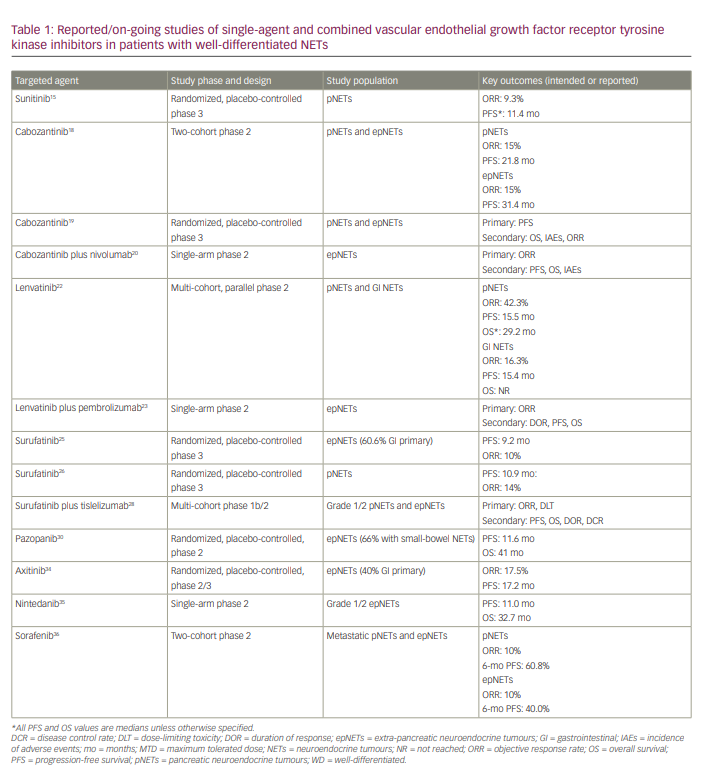
Mechanistic target of rapamycin inhibitors
The mTOR pathway is key in regulating cellular proliferation and growth and is implicated in the proliferation of NET cells.37 NETs tend to have mutations or aberrant expressions of this pathway, which leads to overexpression of mTOR and its downstream molecules and, consequently, uncontrolled tumour growth. Due to its widespread expression across all NETs, regardless of primary tumour site, mTOR provides a viable therapeutic target.1,6,7 In the next section, we discuss mTOR inhibitors.
Everolimus
Everolimus (RAD001) is an mTOR inhibitor. Its activity was first evaluated in a phase 2 trial that included patients with advanced grade 1/2 epNETs and pNETs (n=30 each).38 Patients received either 5 mg or 10 mg doses of everolimus in combination with monthly octreotide. Patients demonstrated an ORR of 22% and a disease control rate of 92%. Median PFS was 60 weeks, and median OS was not reached. The most common grade 3/4 AEs were hypophosphataemia (11%), fatigue (11%) and diarrhoea (11%). Both 5 mg and 10 mg doses were well tolerated.38
The RADIANT-2 (A randomized, double-blind placebo-controlled, multicenter phase III study in patients with advanced carcinoid tumor receiving octreotide depot and everolimus 10 mg/Day or octreotide depot and placebo; ClinicalTrials.gov: NCT00412061) study was a placebo-controlled phase 3 trial in which 429 patients with grade 1/2 advanced NETs (94% epNETs; >50% mid-gut NETs) with carcinoid syndrome were randomized to receive octreotide ± everolimus.39 The median PFS for the everolimus group was 16.4 months and 11.3 months for the placebo group (HR: 0.77; 95% CI: 0.59–1.00; p=0.026). The most common grade 3/4 AEs in the everolimus group were stomatitis (7%), fatigue (7%), diarrhoea (6%) and hyperglycaemia (5%). The primary limitation of this study was that, by a local investigator review, the study exceeded its upper bound α for significance (p=0.025), while by a central investigator review, the study met its primary PFS endpoint (p=0.018). Despite the statistical nuances of the study, the PFS in the combination arm was markedly longer than in the octreotide alone arm.39 As such, in clinical practice, everolimus is often added to octreotide in patients with functional NETs who have progressed on the somatostatin analogue alone.
The RADIANT-3 trial was a randomized phase 3 study (A randomized double-blind phase III study of RAD001 10 mg/d plus best supportive care versus placebo plus best supportive care in the treatment of patients with advanced pancreatic neuroendocrine tumor [NET]; ClinicalTrials.gov identifier: NCT00510068) that enrolled 410 patients who had progressive, advanced, grade 1/2 pNETs.40 Patients were randomized 1:1 to everolimus 10 mg daily or placebo in addition to best supportive care. The median PFS was 11.0 months with everolimus compared with 4.6 months with placebo (HR: 0.35; 95% CI: 0.27–0.45; p<0.001). Treatment-related AEs in the experimental arm were mostly grade 1/2 and included stomatitis (64%), rash (49%), diarrhoea (34%), fatigue (31%) and infections (23%), which were primarily upper respiratory. The most common grade 3/4 AEs in the experimental arm included anaemia (6%) and hyperglycaemia (5%).40 Based on these study findings, everolimus was approved by the FDA in 2011 for the treatment of advanced pNETs.41
In the phase 3 RADIANT-4 trial (A randomized, double-blind, multicenter, phase III study of everolimus [RAD001] plus best supportive care versus placebo plus best supportive care in the treatment of patients with advanced NET of GI or Lung Origin; ClinicalTrials.gov identifier: NCT01524783), 302 patients with advanced, progressive, well-differentiated, non-functional epNETs (30% with mid-gut NETs) were randomized 2:1 to receive everolimus 10 mg daily or placebo, in addition to supportive care.42 The median PFS was 11.0 months in the everolimus group and 3.9 months in the placebo group (HR: 0.48; 95% CI: 0.35–0.67; p<0.00001). Grade 3/4 AEs were infrequent in the everolimus arm and similar to those noted in the RADIANT-3 study.42 The most common grade 3/4 AEs included stomatitis (9%), diarrhoea (7%), infections (7%), anaemia (4%), fatigue (3%) and hyperglycaemia (3%). This study established the anti-tumour activity of everolimus in epNETs and led to its approval in lung and gastrointestinal epNETs.43
Unfortunately, despite its initial anti-tumour activity, everolimus resistance is largely inevitable, leading to disease progression.40,42 To overcome this, inhibitors of p21-activated kinase 4 (PAK4) and the nicotinamide adenine dinucleotide biosynthesis enzyme nicotinamide phosphoribosyltransferase (NAMPT) are being developed. PAK4 and NAMPT are mTOR regulators, which are aberrantly overexpressed in preclinical NET models and lead to everolimus resistance.44 KPT-9274 is a PAK4-NAMPT dual inhibitor that, when administered at 150 mg/kg with everolimus (2.5 mg/kg), worked synergistically to suppress two pNET-derived xenografts.44 Enhanced understanding of tumour biology and molecular behaviour of NETs may lead to the development of combinations to overcome tumour resistance mechanisms.
Emerging targets
Hypoxia inducible factor 2α antagonist
Hypoxia inducible factors (HIF) play a key role in cellular metabolism, and dysregulation of these proteins drives tumourigenesis.45 Tumour hypoxia and overexpression of HIF-1α and HIF-2α, as well as the upregulation of genes directly related to HIF signalling, have been demonstrated in NETs.45 Belzutifan (MK-6482), a small-molecule inhibitor of HIF-2α, has been recently approved for treating VHL disease-associated tumours based on a phase 2 study (An open-label phase 2 study to evaluate PT2977 for the Treatment of Von Hippel Lindau disease-associated renal cell carcinoma; ClinicalTrials.gov identifier: NCT03401788) that showed an ORR of 49% in VHL-associated clear cell renal cell carcinoma.46,47 In this study, among the 22 patients with concomitant non-metastatic pNETs, 90.9% achieved a response.
In light of this striking activity, a subsequent phase 2 trial has been initiated in patients with pancreatic NETs (A phase 2 study to evaluate the efficacy and safety of belzutifan [MK-6482, Formerly PT2977] monotherapy in participants with advanced pheochromocytoma/paraganglioma [PPGL] or pancreatic neuroendocrine tumor [pNET]; ClinicalTrials.gov identifier: NCT04924075).48 This on-going trial is evaluating the efficacy and safety of belzutifan monotherapy in patients with advanced phaeochromocytoma/paraganglioma or pNETs. Belzutifan is being administered at a daily oral dose of 120 mg until progressive disease or unacceptable toxicity. A total of 140 patients are planned for enrolment, and the primary endpoint is ORR.
Antibody–drug conjugates
Somatostatin receptor 2 (SSTR2) is highly overexpressed on NET cells, and SSTR peptide agonists are rapidly internalized in these tumour cells after binding to the receptor.49 Therefore, SSTR agonists can be coupled with cytotoxic drugs, just as they can be coupled with radionuclides, to deliver a lethal dose of DNA-damaging therapy directly to SSTR2-expressing cells. PEN-221 is one such antibody–drug conjugate consisting of the microtubule-targeting agent mertansine (DM1) linked to the C-terminal side-chain of Tyr3-octreotate, which has demonstrated tumour regression in SSTR2-expressing xenograft mouse models. This miniaturized peptide–drug conjugate undergoes internalization and leads to the receptor-dependent inhibition of cellular proliferation.50
A phase 2 study (A phase 1/2a, open-label multicenter study to assess the safety, tolerability, pharmacokinetics, and preliminary antitumor activity of PEN-221 in patients with somatostatin receptor 2 expressing advanced cancers, including gastroenteropancreatic or lung or thymus or other neuroendocrine tumors or small cell lung cancer or large cell neuroendocrine carcinoma of the lung; ClinicalTrials.gov identifier: NCT02936323) tested the safety and efficacy of PEN-221 in 32 patients with advanced well-differentiated epNETs (primarily mid-gut NETs).51 PEN-221 was administered once every 3 weeks intravenously. The maximum tolerated dose was deemed to be 8.8 mg/m2. Of the 26 patients who were evaluable for response, 23 (88.5%) demonstrated stable disease, and target-lesion shrinkage was observed in 10 (38.5%) patients. The median PFS was 9 months. The most frequent (≥20% patients) AEs of any grade were fatigue (39%), nausea (38%), diarrhoea (35%), decreased appetite (30%), infusion reaction (24%), transaminitis (24%) and peripheral neuropathy (21%). The most common grade 3/4 AEs were fatigue (7.6%), transaminitis (7.6%) and peripheral neuropathy (3%).51 Based on the encouraging findings of this phase 2 study, a randomized trial of PEN-221 in patients with epNETs (mid-gut NETs) is being planned.
Cyclin-dependent kinase 4/6 inhibitors
In a genomic analysis of pNET samples, gene amplification and overexpression of CDK4, CDK6 and cyclin D1 proteins, along with increased levels of phosphorylated retinoblastoma protein (Rb1), were observed.52 In addition, CDK4/6 inhibitors suppress tumour growth in the QGP1 pNET xenograft mouse model.52
Results from this preclinical work formed the basis of the open-label, phase 2 PALBONET trial (A phase 1/2a, open-label multicenter study to assess the safety, tolerability, pharmacokinetics, and preliminary antitumor activity of PEN-221 in patients with somatostatin receptor 2 expressing advanced cancers, including gastroenteropancreatic or lung or thymus or other neuroendocrine tumors or small cell lung cancer or large cell neuroendocrine carcinoma of the lung; ClinicalTrials.gov: NCT02334527), which included 21 patients with metastatic grade 1/2 pNETs.53 Patients had previously received a median of 3 prior lines of systemic therapy. Palbociclib, an oral CDK4/6 inhibitor, was administered at a dose of 125 mg daily for 21 of 28 days until disease progression or unacceptable toxicity. Of the 19 evaluable patients, 11 had stable disease ,and 8 had disease progression. The ORR was 0%. The median PFS was 2.6 months, and the median OS was 18.7 months. Asthenia (76.2%), neutropenia (42.9%), diarrhoea (33.3%) and nausea (33.3%) were the most frequently noted AEs. Grade 3/4 neutropenia and thrombocytopenia were observed in 23.8% and 9.5% patients, respectively. Unfortunately, despite encouraging preclinical data, palbociclib failed to demonstrate clinically meaningful responses in patients with pNETs.53
Another pilot study recruited 20 patients with advanced progressive NETs (50% pNETs) to evaluate the efficacy of the CDK4/6 inhibitor ribociclib.54 Ribociclib was administered at 600 mg daily for 3 weeks in a 4-week cycle. Similar to patients treated with palbociclib, no patients treated with ribociclib demonstrated objective response. The median PFS was 10.4 months. There were no grade 4 AEs, but the most common grade 3 AE was neutropenia.54 Another phase 2 trial is currently on-going to test the efficacy and safety of ribociclib 200 mg daily in combination with everolimus 5 mg daily in patients with epNETs (A phase II trial of LEE011 in combination with everolimus in the treatment of advanced well differentiated neuroendocrine tumors of foregut origin; ClinicalTrials.gov identifier: NCT03070301).55
Bispecific T-cell engagers
Apart from the development of the antibody–drug conjugate PEN-221, SSTR2 overexpression on NETs is being targeted in additional ways. Tidutamab (XmAb18087), a humanized, anti-SSTR2 × anti-CD3 bispecific antibody, concomitantly binds to cytotoxic T cells and SSTR2-positive tumour cells, thereby directing cytotoxic T cells to SSTR2-expressing tumour cells.56
A phase 1a/1b trial is currently on-going to evaluate the safety and define the maximum tolerated dose of tidutamab, though preliminary results are available.56,57 The drug is administered as weekly infusions in 28-day cycles to patients with advanced NETs and gastrointestinal stromal tumours. So far, 42 patients with NETs (predominantly pNETs and treated with a median of 4 prior lines of therapy) have been enrolled.57 Grade 3 treatment-related AEs included nausea and vomiting (14.6%), diarrhea (9.8%), anemia (7.3%) and cytokine release syndrome (4.9%).57 The best response observed in patients was disease control, which was observed in 55% of response-evaluable patients. The safe dose range has been established at 0.3–1 μg/kg.57 It remains to be seen how this drug will be further developed given its somewhat modest anti-tumour activity in the monotherapy setting.
Neurotrophic tyrosine receptor kinase inhibitors
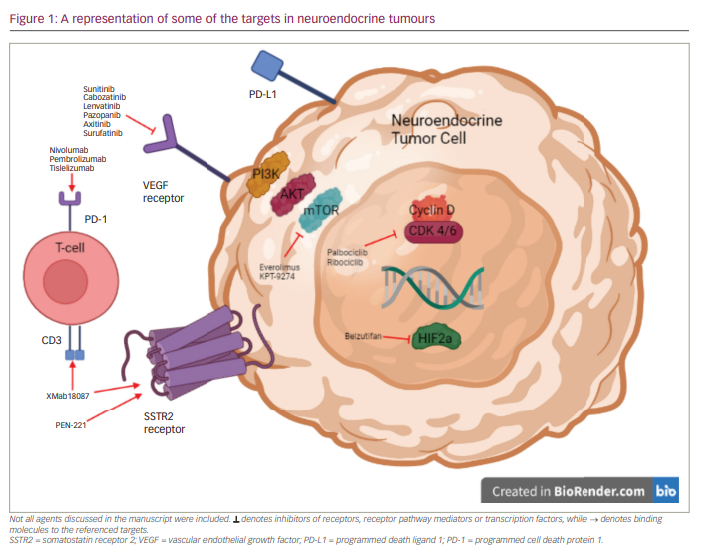
Neurotrophic tyrosine receptor kinase (NTRK) gene fusions involving either NTRK1, NTRK2 or NTRK3 are detected in multiple tumour types and act as driver mutations.58 Comprehensive genomic profiling of >60,000 NET cases from diverse anatomic sites – including small intestine, pancreas and lung – identified 2,417 patients (fusion frequency approximately -0.3%) with these aberrations.59 A single case has been reported of a patient with metastatic, well-differentiated, grade 1 NET (most likely of small intestine origin) with a ETV6:NTRK3 fusion who had a remarkable response to entrectinib, na NTRK fusion inhibitor.60 Even though these mutations are rare, they are actionable when detected. Some of the drugs discussed, along with their targets, are summarized in Figure 1.
Conclusions
An improved understanding of the tumour biology of NETs has more treatment options being developed for patients. Despite these efforts, managing patients with NETs remains a clinical challenge. Results from on-going definitive studies of VEGF RTKIs may lead to new agents being approved; however, sequencing these agents in the backdrop of PRRT will be another therapeutic dilemma. Personalizing treatments for patients based on individual NET biology, overcoming resistance to targeted agents, discovering further pathways implicated in pathogenesis and defining treatment algorithms (based upon primary tumour origin and tumour grade) will evolve care for patients with well-differentiated NETs.61