Prostate cancer is the second most common cause of cancer mortality in men, with most deaths occurring as a result of metastatic disease that has become resistant to androgen deprivation therapy (ADT).1 Most recently, immunotherapy, in the form of immune checkpoint inhibitors, has been successful in the treatment of a variety of advanced common solid tumours, including melanoma, lung, kidney and bladder cancer.2 The key benefit, highly durable response, has resulted in these drugs being transformative for the lives of some patients whose prognosis might otherwise have been bleak. Only a minority attain such benefits, and unpredictable toxicity, acquired resistance and high cost burden remain key challenges in its use across the spectrum of advanced solid tumours.
Advanced prostate cancer
Several drugs have been developed to treat metastatic castration- resistant prostate cancer (mCRPC), including sipuleucel-T, radium-223, docetaxel, cabazitaxel, abiraterone and enzalutamide.3 However, median survival gains with each of these agents are in the range of 3–5 months, and few patients experience durable disease control. In recent years, the treatment of metastatic hormone-sensitive prostate cancer (mHSPC) has evolved, with the earlier use of docetaxel, abiraterone, enzalutamide or apalutamide, all leading to further improvements in survival when given prior to the onset of castration resistance.4–9 Despite several early positive trials, more than a decade later, immunotherapy in the form of sipuleucel-T has only a limited role in the treatment of mCRPC.1,10 This is a review of the existing literature of immunotherapy agents in treating prostate cancer.
Immunotherapy modalities
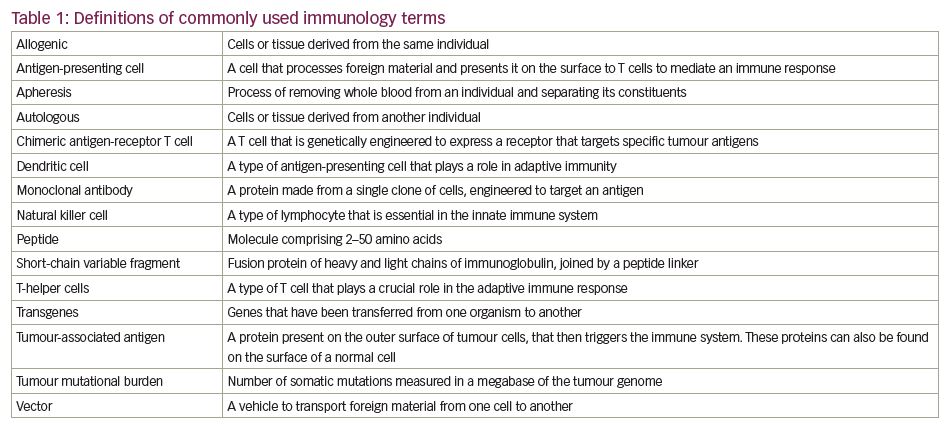
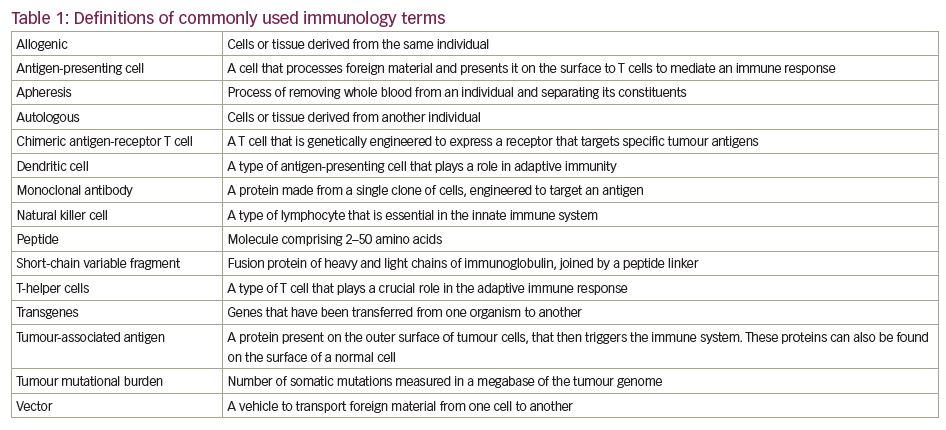
There are a number of ways in which the immune system can be manipulated to target tumours. Vaccines can be derived against causative agents (e.g., human papillomavirus and cervical cancer) or against tumour antigens themselves. The latter can take the form of vaccines derived either directly from peptides, or indirectly through DNA or RNA by targeting either ‘generic’ or personalised tumour antigens.11 Immune checkpoint inhibitors are monoclonal antibodies that target the non-antigen-specific endogenous modulators of the T cell-mediated immune system, thus activating the body’s immune response.11 More recently, therapies have been developed that re-programme cellular immunity to directly target tumour antigens, such as chimeric antigen receptor (CAR) T-cell therapy. See Table 1 for definitions of commonly used immunology terms.
Prostate cancer vaccines
Therapeutic cancer vaccines are one of the earlier forms of immunotherapy studied in prostate cancer and have ongoing interest, partly due to the numerous tumour-associated antigens in the prostate tumour microenvironment, e.g., prostate-specific antigen, prostate-specific membrane antigen, prostatic acid phosphatase and prostate stem cell antigen.12,13 These vaccines induce T-cell responses via different methods. In addition, several trials have shown signals of activity, with one phase III trial demonstrating a survival benefit, leading to drug approval.1
Cell-based vaccines
Cell-based vaccines comprise autologous or allogenic whole cells including antigen-presenting cells and prostate cancer cells that are modified to bear tumour-associated antigens.14 Sipuleucel-T (APC8015) is composed of autologous antigen-presenting cells, extracted from the patient by apheresis, and cultured ex vivo with fusion protein PA2024, which consists of prostatic acid phosphatase linked to granulocyte-macrophage colony-stimulating factor (GM-CSF).1 Prostatic acid phosphatase is expressed in most prostate cancer cells, and sipuleucel-T stimulates a T-cell response to prostatic acid phosphatase.1,15,16 The fresh cellular product is re-infused and the cycle repeated up to a total of three rounds.
Small et al. published the results of a multicentre phase III study of 127 patients with mCRPC in the USA, conducted between 2000 and 2001.15 Patients were randomly assigned in a 2:1 fashion to receive three infusions of sipuleucel-T or placebo intravenously every 2 weeks. There was no statistically significant improvement in the primary endpoint of median time to progression (TTP) (11.7 weeks in the sipuleucel-T arm compared to 10 weeks in the placebo arm; p=0.052, hazard ratio [HR] 1.45, 95% confidence interval [CI] 0.99–2.11).15 However, although not powered to detect a survival difference, survival follow-up was conducted and sipuleucel-T showed a 4.5-month benefit in median overall survival (OS) (25.9 months compared with 21.4 months [p=0.01, HR 1.7, 95% CI 1.13–2.56]).15 Sipuleucel-T retained its survival advantage after adjusting for prognostic factors, i.e., lactate dehydrogenase, prostate-specific antigen, number of bone metastases, body weight and localisation of disease.15 The disparity in results of TTP and median OS might be explained by the time taken for maximum T-cell reactivity when sipuleucel-T is administered, which is thought to be approximately 8–10 weeks.15 It is estimated that patients with mCRPC with asymptomatic bone metastases have a median TTP of about 12 weeks. This would account for the marked initial decline in the TTP Kaplan–Meier curves seen in both patient groups. Furthermore, immunotherapy trials in other tumour types, e.g., lung, head and neck, and urothelial carcinoma, have also reported similar results, with an improvement in OS, but not progression-free survival (PFS), when compared with chemotherapy.17–20 This discrepancy suggests that PFS may be a less useful endpoint in immunotherapy trials given the time to response in the group of patients who do have tumour regression. Rigors, fatigue and pyrexia were the most common adverse events, occurring in >25% of patients.15
Following the results of this preliminary study, Kantoff et al. reported a confirmatory phase III trial with OS as its primary endpoint.1 The IMPACT trial recruited 512 patients between 2003 and 2007, and demonstrated a similar survival advantage (25.8 months versus 21.7 months [p=0.03, HR=0.78, 95% CI 0.61–0.98]) with no difference in TTP.1 This led to approval by the US Food and Drug Administration (FDA) in 2010. However, a subsequent European marketing authorisation was withdrawn due to the absence of a viable European production facility.16 Critics have scrutinised the trial design, commenting on the worse median OS seen in older patients, which had not previously been seen as a prognostic factor in mCRPC patients.21 Furthermore, two-thirds of the harvested cells from patients in the placebo arm were not re-infused, which might have led to lymphodepletion. Cells in the placebo group were incubated without GM-CSF, and at a lower temperature, which may have caused cell death. All these factors may have led to the difference in survival outcomes seen between both groups, and biased the results to favour the sipuleucel-T group.21 Access to this treatment is limited by the necessity to maintain fresh cell cultures throughout, the requirement for proximity to a central production facility, and high cost.22 Some of these issues may be addressed by the VIABLE trial (ClinicalTrials.gov identifier: NCT02111577), assessing a novel autologous dendritic cell-based vaccine that can be stored frozen, and re-infused in aliquots to avoid the need for repeated apheresis and shipment of fresh product. The vaccine studied in this trial, an autologous dendritic cell vaccination (DCVAC/PCa), is an autologous dendritic cell-based vaccine composed of activated dendritic cells pulsed with killed prostate cancer cells from the LNCaP cell line. This trial has completed accrual and is in follow-up. Other formulations of dendritic-cell vaccines pulsed with tumour-associated antigens are being developed.
The GVAX® vaccine (Cell Genesys Inc., San Francisco, CA, USA) is another example of a cell-based vaccine consisting of irradiated hormone-sensitive (LNCaP) and hormone-resistant (PC-3) cancer cell lines that constitutively express GM-CSF.23,24 Although earlier trials of GVAX were promising, two phase III trials were terminated early because of lack of survival benefit (NCT00089856, NCT00133224). VITAL-1 compared GVAX to docetaxel in patients with mCRPC and VITAL-2 randomised patients to the combination of GVAX and docetaxel or docetaxel alone.23,24 The latter study was terminated early due to safety concerns, which led to an early analysis of VITAL-1, indicating futility and, therefore, trial closure.23,24 Reasons for lack of efficacy may have been due to the trial design, which included sub-optimal dosing of both agents and the comparison arm being an active treatment rather than placebo.25
Vector-based vaccines
Genetically engineered nucleic acids that encode specific tumour-associated antigens transmitted by vectors such as viruses or bacterial plasmids have also been studied.26 An immune response is generated by the infection of epithelial cells, which lyse and liberate antigens that are then taken up by antigen-presenting cells and presented to T cells.26 PROSTVAC® (developed by the National Cancer Institute and licensed to BN Immunotherapeutics, Mountain View, CA, USA) was the most developed example using prostate-specific antigens to stimulate a T-cell response and is composed of two live poxvirus-based vectors (rilimogene galvacirepvec and rilimogene glafolivec).27,28 Both of these contain transgenes for prostate-specific antigens and a TRIad of
CO-stimulatory Molecules (TRICOM: B7-1, leucocyte function-associated antigen-3 and intercellular adhesion molecule-1).27,28 After encouraging results in phase II,28,29 the phase III trial, PROSPECT, was stopped early due to futility.30 Between 2011–2015, 1,286 patients were randomised between PROSTVAC, PROSTVAC plus GM-CSF, or placebo, with median OS being 34.4, 33.2 and 34.3 months in the three arms, respectively.30 Another such virus-based vaccine is Ad5 prostate-specific antigen, an adenovirus type 5-based vaccine studied in patients with mCRPC in a phase I trial and deemed to be safe with 55% of patients surviving longer than predicted by the Halabi nomogram.31 Bacteria can also be used as vectors, and one such example of a bacteria-based vaccine utilises Listeria monocytogenes, an intracellular pathogen, which is engineered to express tumour-associated antigens in order to induce an immune response.32 Clinical trials assessing this are currently underway (NCT02625857, NCT02325557).
DNA- and RNA-based vaccines
An immune response can be evoked by DNA-based vaccines through the incorporation of DNA by the host cell and recruitment of antigen-presenting cells. These vaccines can be designed to exploit tumour-associated antigens in order to activate tumour antigen-specific T cells.33 In a phase II trial of pTVG-HP, a plasmid DNA that encodes prostatic acid phosphatase, 99 patients with non-metastatic hormone-sensitive prostate cancer were randomised to pTVG-HP with GM-CSF or GM-CSF alone.34 Two-year metastases-free survival was not different between study arms (41.8% versus 42.3%; p=0.97) nor was the change in prostate-specific antigen doubling time.34 In advanced prostate cancer, pTVG-HP is being assessed as monotherapy or in combination with other immunotherapy agents (NCT04090528). Another method of utilising DNA-based vaccines is using neo-antigens instead of tumour-associated antigens to stimulate T cells.35 Dendritic and peptide neo-antigen DNA vaccines are already being assessed in early phase clinical trials.35
RNA-based vaccines have also been studied. CV9104, a messenger RNA vaccine encoding several tumour-associated antigens was assessed in chemotherapy-naïve patients with mCRPC with no, or minimal, symptoms.36 The phase IIb study randomised 197 patients to intradermal CV9104 or placebo.36 There was no significant difference in OS, radiographic PFS (rPFS) or time to symptomatic progression.36
Peptide-based vaccines
These vaccines use tumour-specific peptides to induce cytotoxic T-lymphocyte activation. Noguchi et al. treated 100 patients with progressive mCRPC with a personalised peptide vaccine.37 It was deemed to be well tolerated and immunoglobulin G and T-cell responses were strongly correlated with prostate-specific antigen doubling time and OS on multivariate analysis.37 Another personalised peptide vaccine was also assessed, combined with estramustine phosphate versus standard-dose estramustine phosphate in patients with human leukocyte antigen (HLA)-A2 or HLA-A24-positive mCRPC.38 The personalised peptide vaccine plus low-dose estramustine phosphate cohort of patients had lower rates of prostate-specific antigen progression. OS also favoured the personalised peptide vaccine plus low-dose estramustine phosphate group (p=0.0328, HR 0.3, 95% CI 0.1–0.91).38 Low-dose estramustine phosphate was used due to proven activity in mCRPC and minimal effect on immunosuppression. Another example is the phase I study of reverse transcriptase subunit of telomerase (hTERT) peptide vaccine UV1 in 22 patients with newly diagnosed mHSPC in addition to androgen deprivation therapy, which showed an acceptable safety profile.39 Other examples of peptide-based vaccines studied in prostate cancer are ITK-1 and GX301.40,41
Non-vaccine approaches to prostate cancer immunotherapy
Chimeric antigen receptor T-cell therapy
CAR T-cell therapy has made significant progress in the treatment of haematological malignancies and is being explored in the treatment of solid tumours including prostate cancer.42 Autologous T cells are engineered ex vivo and comprise an extracellular domain (single-chain fragment variable; scFV) involved in identifying tumour-associated antigens, a transmembrane domain that links the scFV to a T cell, and an intracellular zone that comprises the immune receptor tyrosine-based activation motif that is important in signal transduction and T cell activation.42 Limited clinical work on CAR T cells has been published in the treatment of solid organ malignancies, but it has been shown to be deliverable in patients with prostate cancer.43 Numerous studies are currently ongoing, most of which are assessing CAR T cells directed at prostate-specific membrane antigen (NCT04227275, NCT04249947). See Figure 1 for non-vaccine approaches in treating malignancies.
Bispecific T-cell engager therapy
Another form of immunotherapy using bispecific T-cell engagers (BiTEs) has been explored recently. BiTEs mount a T-cell response by using two linked scFVs from two different antibodies, one targeting a specific antigen on the surface of cancer cells and the other binding to a cell surface molecule on T cells, e.g., CD3.42 Pre-clinical models have shown tumour regression, one example being AMG-160, a fully human BiTE targeting prostate-specific membrane antigens in prostate cancer cells and CD3 in T cells.44 Another BiTE, AMG-212 (pasotuxizumab), has shown clinical activity and an acceptable safety profile in a phase I study of 16 patients with mCRPC.45
Immune checkpoint inhibitors
Cytotoxic T-lymphocyte antigen-4 inhibitors
Checkpoint inhibitors are approved in numerous solid malignancies with cytotoxic T-lymphocyte antigen-4 (CTLA-4), a vital checkpoint and negative regulator of T cells, first discovered in the 1990s.46 Co-stimulatory molecules are needed for immune activation, and the interaction of B7 on the antigen-presenting cell with CTLA-4 on the T cell leads to downregulation.47 Ipilimumab, a CTLA-4 inhibitory antibody, promotes T-cell activation and infiltration. Prostate cancer was one of the first diseases where this agent was tested in pre-clinical models.47 Small et al. conducted a pilot study of ipilimumab in patients with mCRPC showing a prostate-specific antigen decline of ≥50% in 2/14 patients.48
At this time, there was interest in the combination of radiotherapy and immunotherapy (potentially enhancing the abscopal effect, where radiotherapy at one site induces response at distant sites by a presumed immune mechanism). Pre-clinical models demonstrated enhanced anti-tumour activity of checkpoint inhibitors with radiotherapy by releasing tumour antigens and promoting local infiltration of immune cells into the tumour.49 In a phase II trial by Slovin et al., 50 patients were treated with one of the following regimens: ipilimumab 10 mg/kg every 3 weeks for four cycles, or radiotherapy to up to three bone sites prior to ipilimumab administration.50 Some of these patients were chemotherapy naïve. Almost 25% of patients had a ≥50% prostate-specific antigen decline.50 This led to two placebo-controlled phase III trials assessing ipilimumab in patients with mCRPC. The first, published in 2014, included 799 patients who, between 2009–2012, had progressed during or within 6 months of receiving the last cycle of chemotherapy.51 All were given a single fraction of radiotherapy to any site of metastasis within 2 days of starting placebo or ipilimumab 10 mg/kg every 3 weeks for four cycles, followed by maintenance every 3 months.51 The median OS was 10 months versus 11.2 months for placebo and ipilimumab, respectively (HR 0.85, 95% CI 0.72–1.00, p=0.053).51 Despite this, a post-hoc subgroup analysis showed a significant survival benefit favouring ipilimumab in patients with favourable prognostic features (alkaline phosphatase <1.5 x upper limit of normal, haemoglobin <110 g/L and no visceral metastases).51 In this subgroup, median OS in the ipilimumab arm was 22.7 months versus 15.8 months with placebo (HR=0.62, 95% CI 0.45–0.86, p=0.0038).51 In the second trial, ipilimumab was assessed in asymptomatic or minimally symptomatic patients with chemotherapy-naïve mCRPC with a 2:1 randomisation to ipilimumab or placebo, but without the use of radiotherapy.52 Six hundred and two patients were randomised between 2010–2015 and again, no improvement in OS was seen (28.7 months in the ipilimumab arm versus 29.7 months in the placebo arm [HR 1.11, 95% CI 0.88–1.39, p=0.3667]).52 However, there was an increase in PFS and prostate-specific antigen response rate. Despite the overall negative findings, it has been reported that two patients in these phase III trials have had a sustained complete response more than 4 years after randomisation.52
Programmed death 1/programmed death-ligand 1 inhibitors
Topalian et al. first showed promising activity of nivolumab in patients with melanoma, non-small-cell lung cancer (NSCLC) and renal cell cancer in 2012.53 Seventeen patients with castration-resistant prostate cancer were included in the study with no objective responses seen. The KEYNOTE-028 basket trial showed a 17.4% objective response rate (ORR), with 34.8% achieving stable disease and a median duration of response of 13.5 months in patients with mCRPC receiving pembrolizumab 10 mg/kg administered every 2 weeks.54 Of the 23 patients included, 73.9% had received at least two prior therapies for metastatic disease.54 Given these positive results, KEYNOTE-199 was a multi-cohort phase II trial in patients with mCRPC. The first cohorts to be reported included patients exposed to ≥1 prior targeted endocrine therapy and 1–2 prior chemotherapy regimens, including docetaxel, who were grouped in three separate cohorts. Cohort 1 were programmed death-ligand 1 (PD-L1)-positive, cohort 2 were PD-L1-negative, and cohort 3 had detectable bone metastases regardless of PD-L1 status.56 Cohorts 1 and 2 had response evaluation criteria in solid tumours (RECIST) measurable disease. A total of 258 patients were enrolled, with an ORR of 5% and 3% in cohorts 1 and 2, respectively.55 Two patients in cohort 1 achieved a complete response.55 Treatment-related adverse events (TRAEs) occurred in 60% of patients, with 15% of the total cohort experiencing grade 3–5 TRAEs.55 Median OS was 9.5, 7.9 and 14.1 months in each of the three cohorts, respectively.55 These results showed that PD-L1 was not a positive predictive biomarker for response to pembrolizumab, nor were defects in DNA damage repair (DDR) genes.55 Paradoxically, there was a higher response rate in patients in whom DDR defects were absent.56 In addition to nivolumab and pembrolizumab, the efficacy of avelumab has also been assessed in the expansion cohort of the phase I JAVELIN trial. Patients who had progressed on an androgen receptor antagonist received avelumab 10 mg/kg every 2 weeks.57 There were no objective responses seen, and 7/18 patients had stable disease >24 weeks after treatment.57
Combination regimen
Several combination regimens have been explored to enhance anti-tumour activity by combining two immunotherapy agents or by the addition of chemotherapy, second-generation anti-hormonal agents, or poly(adenosine diphosphate-ribose) polymerase (PARP)-inhibitors to a checkpoint inhibitor.
Vaccine-based therapies plus CTLA-4 inhibitors: PROSTVAC has been used in combination with ipilimumab in patients with mCRPC, with a phase I trial showing a median OS of 31.6 months, and 14/24 chemotherapy-naïve patients having a prostate-specific antigen decline.58 Trials combining vaccine-based therapies and checkpoint inhibitors or chemotherapy are ongoing.
PD-L1 plus CTLA-4 inhibitors: Dual checkpoint inhibitor use, e.g., nivolumab and ipilimumab, has conferred a survival benefit in patients with melanoma, renal cell cancer and NSCLC.59–61 CheckMate-650 assessed this combination and showed an ORR of 26% in 78 patients with mCRPC who had progressed after second-generation anti-hormonal therapy, and 10% in those who had progressive disease after taxane-based chemotherapy.62 Response rates were higher in patients with PD-L1 expression ≥1%, presence of DDR defects, homologous recombinant defects or above median tumour mutational burden.62 Of the 62 patients with evaluable disease, four patients experienced a complete response, and durable responses were observed. The main limitation, as demonstrated in previous combined checkpoint inhibitor trials, was toxicity, with only half of the patients receiving all four cycles of ipilimumab and nivolumab, and grade 3–5 adverse event rates in the order of 40–53% with one-third of the total number of patients having to discontinue study treatment.62 Furthermore, four patients had TRAEs leading to death. NEPTUNES, a phase II trial assessing nivolumab and ipilimumab in the treatment of patients with immunosignature positive mCRPC (NCT03061539), is currently ongoing.
PARP inhibitor plus PD-L1 inhibitor: Pre-clinical models with DDR defects are characterised by immune cell infiltration. PARP inhibition, which leads to accumulation of DNA mutations, attracts immune cells to the tumour site.63 Since DDR defects lead to genomic instability and higher tumour mutational burden, a rationale exists for the combined use of checkpoint and PARP inhibitors.64 Karzai et al. assessed the combination of durvalumab and olaparib in 17 patients with mCRPC who had received prior enzalutamide and/or abiraterone.65 This demonstrated a median rPFS of 16.1 months with a 12-month rPFS rate of 51.5%, and median PFS of 7.8 months.65 The KEYNOTE-365 trial of pembrolizumab and olaparib in 41 docetaxel pre-treated patients showed a 7% (2/28) confirmed ORR and 32% (9/28) disease control rate in those with measurable disease.66 None of these patients had homologous recombinant defects and 27% were PD-L1 positive. This trial is also assessing docetaxel or enzalutamide in combination with pembrolizumab in two separate cohorts. A similar phase II study (CheckMate 9KD) is being conducted using nivolumab with similar regimens (rucaparib in place of olaparib).67 Preliminary results in the nivolumab and docetaxel arm showed an ORR of 36.8%, with one complete response.67 The phase III CheckMate 7DX assessing this combination in patients with mCRPC who have been previously treated with second-generation anti-hormonal agents, is ongoing (NCT04100018).
Androgen receptor-targeted therapy combinations: It has been shown that patients progressing on enzalutamide had significantly increased PD-L1/2-positive dendritic cells and T cells compared with enzalutamide-naïve patients or those responding to treatment; in addition, these patients are sensitised to T-cell mediated lysis.68,69 This supported the idea that the lack of significant response seen in patients with prostate cancer, to anti-programmed cell death protein 1 (PD-1) therapy, may be due to the lack of PD-L1 expression,68 and created the platform to launch early studies in which pembrolizumab was added into the treatment regimen of chemotherapy-naïve patients who had progressed on enzalutamide.70 Partial responses were seen in 25% (3/12) of patients, and median OS was 22.2 months. No predictive biomarkers were elucidated.71 The results of cohort C of the KEYNOTE-365 trial were reported in 2019; 69 patients were enrolled and the ORR was 20% (5/25).72 Recently, results were presented for cohorts 4 and 5 of the KEYNOTE-199 trial, which included patients who were progressing on enzalutamide as their only prior therapy for mCRPC, and showed an ORR of 12% (10/12) in cohort 4, with 2 complete responses.55 Sixty per cent of responses were ongoing at 6 months. Ongoing treatment effect was observed
post-discontinuation, and disease control rates for all patients were 51% in both cohorts.55 In enzalutamide-resistant patients, it appears that adding pembrolizumab, rather than switching to pembrolizumab monotherapy, confers a higher response rate. KEYNOTE-641, the phase III trial assessing this combination, is currently recruiting (NCT03834493). Another phase III trial, IMbassador250, evaluated atezolizumab in combination with enzalutamide in patients with mCRPC after failure of a second-generation anti-hormonal agent and failure of, ineligibility for, or refusal of a taxane chemotherapy. There were no differences found in rPFS or OS and the trial was terminated early.73
Limitations of immunotherapy
There exist plenty of rationales for the use of immunotherapy in treating prostate cancer, and numerous studies have shown response. However, results to date have not mirrored the positive findings seen in other tumours, and several proposed theories may explain this. Prostate cancer has been described as a ‘cold tumour’. The prostate cancer tumour microenvironment is immunosuppressive, with downregulation of tumour immunity due to recruitment of regulatory T cells and Th17, low cytolytic activity of natural killer (NK) cells and higher transforming growth factor- beta levels, which inhibits NK and T cell function.74,75 Prostate cancer is not an immunologic solid tumour compared with other tumour types, and is not known to have a high tumour mutational burden, ranking 20th out of 27 tumour types according to median somatic mutation frequency.76 Specific treatments, such as CAR T-cell therapies, have limitations in treating solid organ tumours such as prostate cancer where metastases are predominantly skeletal, including a lack of penetration to bone, the lack of durability of T cells and the need for a higher concentration of T cells because of their engulfment by the reticuloendothelial system.77 Furthermore, drug delivery with vaccine-based therapies pose logistical and financial challenges and are resource intense.77
Patient selection
The lack of robust anti-tumour activity of immunotherapy agents in prostate cancer necessitates better patient selection based on clinical, pathological and genomic features to clearly identify predictive biomarkers. Existing literature suggests that mismatch repair defects (MMRd) in prostate cancer predict a biochemical response to checkpoint inhibitors.78,79 Among 1,033 patients with prostate cancer undergoing treatment at a single centre, 3.1% had microsatellite-high (MSI-H) or MMRd, with 21.9% of these having a pathogenic germline mutation in a Lynch-syndrome associated gene.78 Although uncommon, the National Comprehensive Cancer Network guidelines suggest testing all patients with prostate cancer, in the context of the FDA approval of pembrolizumab for MSI-H tumours, regardless of histology, based on the KEYNOTE-158 basket trial, which included 233 patients, of which six had prostate cancer.80 Deleterious germline DDR mutations such as BRCA, CHEK2, ATM, RAD51D and PALB2 are seen in 12% of patients with mCRPC.81 Current trials assessing immunotherapy in combination with PARP inhibitors are either enriching for these subgroups, including those with DDR defects, or stratifying for these.79 PD-L1, another well-studied biomarker, is expressed in 8% of primary prostate cancer and 32% in mCRPC samples.82 In the KEYNOTE-199 trial, response was independent of PD-L1 expression in the pembrolizumab-alone arm. However, higher response rates were seen in patients who were PD-L1-positive in the CheckMate-650 trial. Ongoing phase III trials are exploring the predictive properties of this biomarker.
Inactivating cyclin-dependent kinase 12 (CDK12) mutations are found in 6–7% of mCRPC cases.83 In a retrospective study conducted by Antonarakis, 58 men were identified with a somatic loss-of-function CDK12 mutation including 48% with biallelic inactivation.84 Eight men received a PD-1 inhibitor as fourth- to sixth-line treatment for mCRPC, with 38% having a prostate-specific antigen response and median PFS of 6.6 months. In comparison, median PFS for those on first-line, second-generation anti-hormonal agents was 4.3 months, and only 4.0 months for those receiving a taxane agent in the first-line setting. These findings suggest that prostate cancer with loss of function CDK12 mutations are aggressive in nature, with poor response to standard approved therapies of taxane-based chemotherapy and second-generation anti-hormonal agents.83,84 There is promising activity in favour of PD-1 inhibitors in this subgroup.85,86
Other unknown factors in the use of immunotherapy include the optimal timing of administration. There is evidence to suggest, over time, exposure and resistance to second-generation anti-hormonal agents increases PD-L1 expression and hence response to checkpoint inhibitors, but whether these agents should be given pre- or post-chemotherapy in the mCRPC setting, at time of biochemical relapse or even earlier in the neoadjuvant setting are all being explored.68,87 The choice of treatment and combination of agents to use are far from being established. Treatments that can restore NK and T cell function, in addition to trials investigating the combination of immunotherapy with radiotherapy, are required. Not only is it key to understand the reasons for lack of response and resistance to immunotherapy, but selecting fit patients who are suitable for these potentially toxic agents is crucial to reduce the risk of increased morbidity and mortality from immune-related adverse events.
Duration of response
One of the key attractions of immunotherapy – as seen in other solid tumours – has not been the proportion of patients who respond, but the duration of that response, which brings with it the prospect of long-term disease control. To date, relatively scarce data have been reported in prostate cancer, but those that have suggest that duration of response may be shorter than that seen with other therapies in prostate cancer, such as endocrine therapies. The underlying reason for this short duration of response to immunotherapy needs to be understood if novel approaches are to be developed to combat this.
In conclusion, immunotherapy trials in advanced prostate cancer have been ongoing for more than a decade, and it is clear that at least some cases are amenable to an immunotherapeutic approach. However, apart from pembrolizumab approved for MSI-H tumours and sipuleucel-T, no other immunotherapy agents have established a role in treating patients with metastatic prostate cancer, and immunotherapy is yet to have the same impact in prostate cancer, as has been seen in other solid tumours. Key to future success is the development of effective predictive markers for current therapies; in particular, markers of durable response to immune checkpoint inhibitors, but also, through a deeper understanding of biology, to the development of novel immune therapeutic approaches that will bring the profound benefits of immunotherapy currently experienced by the minority to all patients where there is an unmet need. These are all areas of ongoing research, which will endeavour to provide more insight.