Colorectal cancer (CRC) is the third most common cancer worldwide and the second most common cause of cancer-related death,1 with its incidence predicted to continue to rise significantly in the future.2 With the evolution of personalized medicine, genomic profiling is crucial for treatment and monitoring of individuals’ cancer. The increasing availability of novel molecular-targeted therapy demands the utilization of an effective tool to accurately profile patients at a molecular level to guide treatment. In metastatic CRC (mCRC), treatment with epidermal growth factor receptor (EGFR) monoclonal antibodies (cetuximab, panitumumab) for wild-type RAS tumors has a proven survival benefit,3 with RAS molecular testing of tumor tissue at diagnosis now a standard of care.4 Combination BRAF-targeted therapy (vemurafenib + cetuximab + irinotecan or encorafenib + cetuximab + binimetinib) has also been recently approved by US Food and Drug Administration (FDA) and incorporated into the NCCN treatment guideline for BRAFV600E mutant mCRC.5
Circulating cell-free DNA (cfDNA) was first discovered in 1948 by Mandel and Metais,6 but it wasn’t until 1977 that cfDNA was recognized to be at a higher serum concentration in patients with cancer.7 cfDNA is the term for genetic fragments in the form of nucleic acid chains found in bodily fluids including blood, stool, urine, and saliva. In healthy individuals, the majority of cfDNA is derived from hematopoietic cells. In patients with malignancies, a proportion of cfDNA is derived from tumor cells, referred to as circulating tumor DNA (ctDNA). The precise mechanism by which ctDNA enters the bloodstream remains unclear, but tumor DNA is thought to be released from tumor cell necrosis or apoptosis.8 Once in the circulation, ctDNA is cleared rapidly from the bloodstream, with a half-life of approximately 2 hours.9,10 The relative short half-life of ctDNA compared to serum protein biomarker such as carcinoembryonic antigen (CEA; half-life of 7 days), offers a distinct advantage for ctDNA as a real-time dynamic biological marker of tumor burden. Another appealing feature of ctDNA as a cancer biomarker is its exquisite specificity, given tumor-specific genetic abnormalities (point mutations, chromosomal translocation, methylation changes) can be detected in the circulation.
However, ctDNA concentration can be as low as <0.01% of the total cfDNA, and has been shown to correlate with tumor burden.11,12 Until recently, the inability to reliably detect and quantify such a low frequency of tumor DNA variants in a standard blood sample has restricted its clinical application. This is particularly relevant in the setting of early stage disease where the tumor burden is low. This has led to the development of specialized methods for ctDNA analysis, ranging from technologies used for a single mutation to entire genome analysis. The single mutation or limited mutation panel strategy utilizes detection of individual point mutations through polymerase chain reaction (PCR)-based assays, such as droplet digital PCR and beads, emulsion, amplification and magnetics (BEAMing). This strategy offers higher sensitivity than non-targeted approaches, but necessitates prior knowledge of mutation information.13 Conversely, non-targeted approaches such as whole genome sequencing using next generation sequencing (NGS) requires no prior knowledge about the tumor mutation,14 but does require higher concentrations of ctDNA and has a lower sensitivity.12 Recently, target sequencing modified with molecular barcoding to reduce background PCR error rates has successfully improved the sensitivity of ctDNA detection approaching that of PCR-based methods.13,15,16 An in depth review of ctDNA analysis methodologies is beyond the scope of this article. Readers are encouraged to refer to several excellent reviews of this topic in the literature.17–19
The focus of this review will be on the recent advances in the clinical applications of ctDNA in both early-stage and mCRC, and the ongoing and upcoming clinical trials investigating the effectiveness of ctDNA-informed therapeutic strategy.
Clinical applications of ctDNA in metastatic colorectal cancer
Tumor genotyping
Tissue-based genotyping is currently the standard of care for the diagnosis and molecular profiling of CRC.20 While surgically resected tumors generate reliable results, biopsy samples are often of inadequate quantity for analysis and can produce false-negative results, which is particularly pertinent to RAS mutation analysis, given the treatment ramifications.20 Repeated tissue biopsies are also expensive, can be technically difficult with inherent complication risks, and have a slow turnaround time. Moreover, a single lesion biopsy only reflects the genomic profile of the tumor in that space at that particular point in time, and may greatly underrepresent the molecular heterogeneity of resistant clones and mechanisms that would impact response to treatment.21
The genetic and molecular landscape of CRC is inherently complex and heterogeneous. Understanding both intra and intertumoral heterogeneity is clinically important, as they may have implications on treatment options. Intertumoral heterogeneity, meaning tumor cells from different metastatic tumors (for example lung and liver metastases) harboring a different mutation profile, is a well-known concept, having been described across numerous cancer types for many years.22–25 Molecular screening of patients with mCRC for KRAS and NRAS mutations to assess suitability for anti-EGFR therapy is now standard practice. However, RAS wild-type colorectal tumors that respond to anti-EGFR therapy almost always develop resistance within 5–6 months of initiating treatment.26 This acquired resistance has been attributed to emerging mitogen-activated protein kinase (MAPK) pathway mutations that were present in minor tumor subclones prior to initiation of treatment.26 Studies have now shown that tumor cells within the same tumor mass in an individual patient can also carry several different molecular alterations, referred to as intratumoral heterogeneity, contributing to drug resistance.21,23,27–29
Whole genome or exome sequencing of DNA from CRC has characterized a number of recurrent somatically mutated genes, such as APC, TP53, KRAS, NRAS, BRAF, and PIK3CA,30,31 affording the ability to perform targeted panel sequencing for liquid biopsies. Several studies have demonstrated that ctDNA profiling can accurately reproduce the genomic landscape of tumor tissue,21,32–34 and more specifically the detection of RAS mutations by ctDNA analysis has also been validated, demonstrating it to be a viable alternative to tissue-based RAS testing.11,35–42 Furthermore, when compared directly with single lesion biopsy, ctDNA from blood can actually pick up distinct resistance mechanisms and molecular heterogeneity in an individual patient, highlighting the pitfalls of treating based on the genomics of a single lesion.21
In 2014, Bettegowda et al. analyzed ctDNA from 640 patients with various cancer types and stages and found that ctDNA was detected in 82% of metastatic solid tumors outside of the brain, and approximately 50% of early stage cancers, but detection rates varied with specific tumor types.11 Specifically for CRC, ctDNA was detected in 75% of early stage (I–III) and 100% in metastatic disease. In the 206 patients with mCRC, the sensitivity and specificity of KRAS gene mutations was 87.2% and 99.2%, respectively.
In 2018, Bachet et al. performed a large prospective study on treatment-naïve patients using a NGS-based extended RAS panel, but also incorporated the use of methylation CRC markers (WIF1 and NPY) to assess for the presence of ctDNA.40 The overall accuracy of 85% is consistent with previous concordance studies,35,37,42 but improved substantially to 95% (with a sensitivity of 93%) when the cases which lack ctDNA by methylation testing (20%) were excluded. This confirms that the lack of ctDNA accounts for the majority of the false-negative ctDNA RAS results. Of note, the absence of liver metastases and primary resection were the main factors associated with a negative ctDNA. Once the analysis was restricted to patients with liver metastases, the accuracy increased to 97%.40 This study highlights that clinical factors including tumor burden, sites of metastases, and timing of blood collection must be considered when interpreting results of ctDNA testing. Given that the accuracy of ctDNA molecular testing is dependent on the abundance and presence of tumor DNA in the circulation, ctDNA analysis for RAS or other mutation status is best performed in treatment-naïve patients or at the time of disease progression.
Monitoring treatment response and resistance
The current gold standard for assessing initial disease bulk and to define treatment response is the image-based Response Evaluation Criteria in Solid Tumours (RECIST), established in 2000 and updated in 2009.43 The limitations of conventional imaging include poor inter- and intra-observer reproducibility, inability for morphological categorization, radiation toxicity, and the possibility of controversial results leading to overtreatment in some individuals.44
Serum CEA is the most widely used tumor marker in CRC, but has a limited role in assessment of response to therapy. A rise in CEA is detectable in just 30–40% of patients diagnosed with CRC at any time.45 Despite CEA being an independent prognostic factor46 and the fact that elevated CEA at initial presentation of CRC is associated with an adverse outcome,47 the low sensitivity and specificity of CEA has limited its use to that of post-surgery surveillance only.20,48–50
There is a high attrition rate of patients with mCRC receiving sequential lines of treatment, with clinical deterioration being a contributing factor. In a US-based population study of almost 5,000 patients receiving first-line chemotherapy, only 53% went on to receive second-line, 28% received third-line, and only 13% received fourth-line treatment.51 As more active agents become available, it will be increasingly challenging to expose all patients with mCRC to effective therapy. Timely identification of patients who will be resistant to treatment raises the possibility of an earlier switch to an alternative, potentially more effective therapy, minimizing toxicities from ineffective treatment and improving outcome.
A prospective observational study has been conducted involving 53 patients with treatment-naïve mCRC receiving standard first-line chemotherapy investigating the ability of dynamic ctDNA change as an early marker of treatment response to first-line chemotherapy.52 Each patient’s tumor tissue was initially sequenced using a panel of 15 genes frequently mutated in mCRC to identify candidate mutations for ctDNA analysis. For each patient, one tumor mutation was selected to assess the presence and the level of ctDNA in plasma samples using a digital genomic assay termed Safe-SeqS. At least one patient-specific candidate tissue mutation for ctDNA analysis was identified in 98.1% of the tumors. This study showed that ctDNA can be detected in a significantly high proportion of patients (92.3%), with early changes in ctDNA during chemotherapy being associated with a treatment response at first restaging. The median ctDNA fraction dropped significantly from a baseline of 16.24% to 0.54% by the start of cycle 2, but there was no significant early change in CEA levels when taken at the same time-points. ctDNA response, defined as a >10-fold reduction in ctDNA mutant allele fraction from baseline to pre-cycle 2, correlated with CT response at 8–10 weeks (>20% tumor size reduction by RECIST), suggesting that early ctDNA change is predictive of treatment response. ctDNA-based response assessment may potentially complement RECIST assessment, especially in patients with non-measurable disease by RECIST criteria. Interestingly, four patients had a transient ctDNA spike 3 days after chemotherapy was initiated. As three-quarters of them had an excellent response to treatment, this spike could be explained by the rapid release of ctDNA from responsive tumors. The rapidity of correction and absence of spikes in other patients could be the result of the short half-life of ctDNA.
Similarly, for molecularly targeted therapy, Corcoran et al. demonstrated a marked decrease in BRAFV600E levels in ctDNA by week 4 in patients who responded to treatment with combination therapy of BRAF, EGFR, and MEK inhibition (dabrafenib, panitumumab, and trametinib), but less so in the stable or progressive group.53 There was no significant early change in CEA in both the responders and non-responders. In the responders, a rebound in BRAFV600E level at the time of disease progression was observed.
The design of individualized and targeted personalized therapy relies on the ability to predict disease course and evolution. In a prospective phase II study, Khan et al. demonstrated that longitudinal ctDNA measurements combined with mathematical modelling of tumor evolution gave the ability to quantitatively anticipate tumor progression.54 Using a combination of blood ctDNA samples at 4-week intervals, repeat biopsies, and RECIST assessment, it was shown that RAS pathway mutations clonally expand during anti-EGFR therapy, and that approximately half of patients with mCRC considered initially to be KRAS wildtype, actually have RAS mutations and have no benefit from anti-EGFR treatment. Integrating sequential mutation data from regular ctDNA samples and tumor burden (CEA and RECIST) with mathematic modelling allowed accurate and dynamic measurement of resistance leading to the ability to predict the time to progression and overcome patient intervariability. This study suggested that ctDNA analysis could provide more information about RAS mutant clones than tissue biopsies, and once again emphasized the limitation of tissue biopsies’ ability to capture intratumoral heterogeneity.
Tracking molecular mechanisms of resistance to targeted therapies
Acquired resistance is a significant barrier to targeted therapy. ctDNA as an easily accessible source of genomic information allows for serial liquid biopsies to be performed, representing a much easier and safer alternative to repeated tissue biopsies. A mechanism of resistance to anti-EGFR therapy was first described in 2012 by Diaz et al., whereby the emergence of circulating mutant KRAS ctDNA in patients receiving monotherapy with panitumumab was assessed.26 This study found 38% of patients with initial KRAS wild-type tumor developed at least one KRAS mutation at the time or prior to disease progression with panitumumab treatment. A third of these patients were found to have multiple different KRAS mutations. In another study of almost 1,400 patients, Strickler et al. highlighted the extent of tumor heterogeneity in individual patients.34 They observed that, on average, patients had five unique resistance genomic alterations on ctDNA testing following EGFR blockade, with as many as 13 different resistance mechanisms found in a single patient. Of note, only 9% of study patients were found to have a singular resistance mechanism. A similar mechanism of resistance with MAPK reactivation was observed in BRAFV600E mutant mCRC treated with dabrafenib, trametinib, and panitumumab. In initial responders, Corcoran et al. observed the emergence of at least one circulating RAS mutation in almost half the patients (48%) at the time of progression.53 Furthermore, 21% of these patients had multiple subclonal RAS mutations at progression.
Applications of ctDNA in early stage colorectal cancer
Minimal residual disease detection and a marker of recurrence
A significant proportion of patients with early stage solid malignancies will develop recurrence after curative intent surgical resection of their primary tumor. This is mainly due to presence of occult micrometastatic or minimal residual disease (MRD) that cannot be detected by conventional imaging modalities. While clinicopathological characteristics of tumors (e.g. T stage and number of nodes involved) are currently used as indicators of propensity for metastasis or recurrence in patients with early stage cancer, they have remained suboptimal for risk stratification.
CRC remains a major cause of cancer-related death worldwide, accounting for over 850,000 deaths per year.1 The most substantial opportunity for improving CRC survival outcomes is in the setting of early stage disease, where evidence indicates that adjuvant chemotherapy can reduce recurrence and prevent cancer-related death through the eradication of MRD. However, the current treatment approach lacks precision and has quite a modest survival impact, which also comes at significant cost. Specifically only a minority of patients benefit if all are offered adjuvant chemotherapy (0–4% of those with stage II and 15–20% of those with stage III), as the majority are either already cured by surgery or will recur despite the selected adjuvant therapy, due to the failure to eradicate residual disease.55–59 In addition to treatment-related cost, all patients are exposed to the risk of significant treatment-related toxicities. Disappointingly, despite multiple randomized trials involving nearly 15,000 patients, no new approach has demonstrated survival gains in the past 15 years.60–66
A powerful prognostic marker could revolutionize adjuvant therapy by (i) better defining the recurrence risk of individual patients and allowing delivery of a more personalized treatment approach (escalating or de-escalating treatment compared to the current standard stage-based treatment), and (ii) enriching studies of new therapies with high-risk patients, substantially reducing the size and cost of adjuvant therapy trials. A blood-based marker could also be used to monitor the effectiveness of adjuvant therapy, generating an early readout of treatment effect in clinical trials.
After curative intent cancer surgery, the presence of ctDNA being released from occult residual cancer cells, is a direct marker of recurrence risk. In a prospective observational study, Tie et al. investigated the ability of ctDNA to detect MRD in 230 patients with resected stage II colon cancer by using massive parallel sequencing-based Safe-SeqS assays.67 They demonstrated that patients with stage II colon cancer who had ctDNA detected in their plasma 4–10 weeks after surgery, were at very high risk of recurrence when not treated with chemotherapy compared to patients where ctDNA was not detectable postoperatively (estimated 3-year recurrence-free survival [RFS] of 0% compared to 90%; hazard ratio [HR], 18; p<0.001). Importantly, the prognostic value of postoperative ctDNA was independent of known clinicopathological risk features. The strong prognostic impact of post-surgery ctDNA was confirmed in another study involving 159 patients with locally advanced rectal cancer, where patients with detectable ctDNA after surgery had a significantly worse RFS (HR 13; p<0.001).68
The clinical application of ctDNA to predict recurrence was also investigated in stage III colon cancer where all patients are offered adjuvant chemotherapy.69 It was observed that patients with positive ctDNA following surgery, prior to commencement of chemotherapy, had a worse RFS compared with ctDNA-negative patients despite the use of adjuvant chemotherapy (HR 3.7; p=0.0014).
Whilst a highly specific test (specificity approaching 100%), the clinical application of ctDNA in early stage disease is currently limited by the ability to detect minute amounts of ctDNA in circulation, with the sensitivity of postoperative ctDNA in predicting recurrence found to be 48%.67 It is possible that improvement in technology and detection of multiple mutations (rather than just one mutation in our study) in the blood sample could improve the sensitivity.
Monitoring adjuvant treatment efficacy
The aim of adjuvant chemotherapy in early stage (stage II and III) CRC is to eradicate MRD and reduce the risk of recurrence. However, the effectiveness of chemotherapy in individuals is not easily measurable in real-time and the impact of therapy on preventing recurrence cannot be easily quantified. There is growing evidence to suggest measuring serial ctDNA postoperatively, during chemotherapy and after completion of treatment in patients with solid tumors is associated with the risk of recurrence and also can be used as a personalized real-time marker of chemotherapy effectiveness.
Anecdotal evidence reports that patients with stage II colon cancer who had persistently detectable ctDNA after completion of chemotherapy, experienced recurrence; whereas those who eradicated their ctDNA at the completion of adjuvant chemotherapy remained cancer free at their last follow-up.67 In the previously mentioned stage III colon cancer study, patients with persistent or developed detectable ctDNA at the completion of chemotherapy had an estimated 2-year RFS of only 27% compared to 82% in those with undetectable ctDNA (HR 7.1; p<0.001).69 Similar results were observed in another study evaluating the correlation between serial ctDNA and recurrence risk in patients with resectable colorectal liver metastasis where all patients with positive ctDNA post-treatment (surgery and chemotherapy) experienced recurrence.70
Authors’ view on the potential clinical utility of ctDNA
Significant progress has been made towards implementing ctDNA in clinical practice in CRC. In the metastatic setting, non-invasive tumor genotyping has been supported by concordance studies and shown to be useful to study molecular mechanisms of resistance. As a marker of treatment response, ctDNA may complement the use of existing imaging criteria, especially for assessing early response and in the setting of non-measurable disease by RECIST criteria. However, radiological imaging remains the current gold standard and could also provide important clinical information such as tumor location which is not available with ctDNA analysis. How an early signal of treatment response could translate into clinical benefit remains to be seen and should be investigated further.
In early stage disease, ctDNA detection predicts high risk of recurrence and has the potential to improve outcomes in enriched populations to investigate new therapeutic approaches and avoid unnecessary chemotherapy. Whilst ctDNA analysis in early stage disease shows great promise, more robust, prospective randomized trials are necessary to establish the clinical utility of ctDNA before this test can be adopted into the clinic for routine care. Another hurdle to overcome is the range of different assays available to measure and analyze ctDNA, a factor which has currently limited ctDNA to the research setting only. Standardized guidelines for ctDNA collection and analysis with technical improvements in the assays used are crucial to facilitate clinical realization.
Current and upcoming randomized clinical trials in adjuvant setting
The ability of ctDNA to stratify patients into very high-risk and low-risk of recurrence creates an opportunity for ctDNA-guided approaches to adjuvant chemotherapy for patients with early stage cancer. This approach could lead to significant survival gains in preventing recurrence through treatment escalation or novel treatment in ctDNA-positive high-risk populations. In contrast, low-risk patients with undetectable ctDNA, could be given less intense treatment which, in turn, results in lower treatment-related toxicity and cost. Using ctDNA to enrich for high-risk patients in clinical trials can also significantly improve trial efficiency by reducing the sample size needed to demonstrate a meaningful difference in outcome given the high event rate.67 In those where adjuvant chemotherapy failed to eradicate residual disease as indicated by a positive ctDNA, additional novel treatments could be tested.
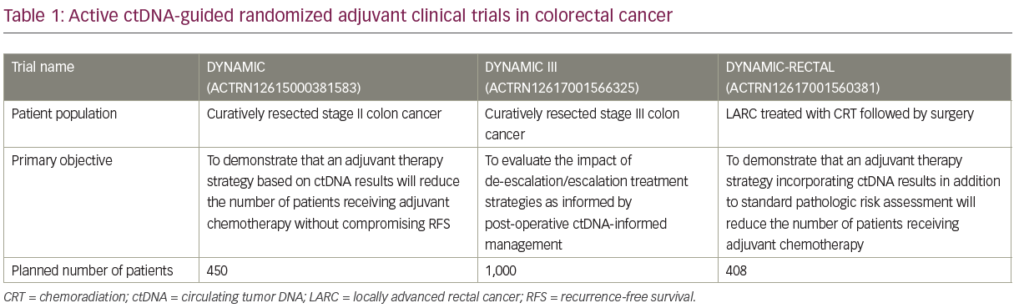
There are currently three active Australasian ctDNA-guided randomized adjuvant clinical trials in early stage CRC (DYNAMIC [stage II], DYNAMIC-III [stage III], and DYNAMIC-RECTAL [locally advanced rectal cancer]; see Table 1). The primary objective of these studies is to evaluate the impact of de-escalation/escalation treatment strategies as informed by post-operative ctDNA-status compared to standard of care (non-ctDNA-guided) approach. In the DYNAMIC (stage II) and DYNAMIC-RECTAL studies, the primary aim is to demonstrate that a ctDNA-guided strategy will reduce the number of patients receiving adjuvant chemotherapy without compromising RFS. In the randomized phase II/III DYNAMIC-III study, ctDNA-positive patients randomized to the biomarker-guided arm will be receiving more intensive chemotherapy including FOLFOXIRI, with the study aiming to demonstrate that this could lead to an improvement in RFS. An NRG/NCTN Oncology phase II/III study under development (CR 1643), aims to enroll patients with resected stage II colon cancer deemed suitable for observation by their physicians; patients will be randomly assigned to observation or adjuvant therapy based on ctDNA results.71 The primary objective of the phase II portion is to evaluate the rate of ctDNA clearance with adjuvant therapy, whereas the primary objective of the phase III portion is to evaluate RFS in ctDNA-positive patients who do and do not receive adjuvant chemotherapy.71
Conclusion
The development of novel molecular targeted treatment options has opened the door for further advancements in the field of personalized oncology in CRC. In mCRC, ctDNA-based tumor genotyping has been supported by concordance studies compared to tissue-based genotyping. ctDNA also has the potential to identify intratumoral heterogeneity and track molecular mechanisms of resistance to targeted therapy. ctDNA changes have also been shown to provide an early signal of treatment response or resistance to chemotherapy and targeted therapy. In early stage CRC, ctDNA detection predicts for very high risk of recurrence, providing opportunities to improve patient outcomes by testing novel treatments in combination with standard chemotherapy in this ctDNA-enriched population. Further prospective clinical trials are critical to support the utility of ctDNA in a defined clinical setting and its impact on patient health outcomes, and we eagerly await the results of these studies. Of equal importance, there should be ongoing effort to standardize pre-analytical and analytical variables for ctDNA analysis including fit-for-purpose collection, processing, and storage procedures, and correlating the performance of various ctDNA assays.