While chemotherapy and radiation remain the mainstays in the initial treatment of Hodgkin’s disease (HD) and non-Hodgkin’s lymphomas (NHL), patients with relapsed disease or those who fail to enter remission are rarely cured using these conventional methods.1,2 Therefore, approaches using adoptive immunotherapies offer attractive alternative options for this subgroup of patients. While infused monoclonal antibodies have been proved to offer success as cancer immunotherapies, it is much more difficult to exploit the equivalent promise of T-cell immunotherapy. This dilemma is two-fold, comprising the significant time required to generate T cells for infusion and the relative lack of persistent in vivo expansion of the adoptively transferred cells, making it difficult to achieve sustained clinical responses. This article will focus on the T-cell therapies available in the treatment of multiple-relapsed or refractory lymphomas, and some of the strategies in place to overcome these obstacles.
Immunotherapeutic Options
Adoptive T-cell immunotherapy approaches to treating lymphomas have been evaluated in both the allogeneic and autologous settings. Durable clinical responses following allogeneic stem cell transplantation (SCT) for lymphomas imply the presence of a graft-versus-lymphoma (GvL) effect. In order to facilitate the generation of specific T-cell therapies for lymphomas, target antigens capable of T-cell recognition must first be identified, such as viral antigens present on tumor cells, or alloantigens, such as minor histocompatibility antigens. Thus, T-cell therapies targeting lymphomas can be divided into two major categories: non-specific T cells, including the use of unmanipulated donor T cells or autologously non-specifically expanded T cells; and antigen-directed cytotoxic T cells, including Epstein-Barr virus (EBV)-specific T cells and chimeric T cells (see Table 1).
Unmanipulated Allogeneic T Cells
(Donor Lymphocyte Infusion)
Perhaps the best known and most widely utilized form of immunotherapy for any malignancy is the use of unmanipulated allogeneic T cells, otherwise known as donor lymphocyte infusions (DLI), following allogeneic hematopoietic SCT. It is now understood that lymphomas can be sensitive to the effects of DLI with varying success rates.3–7 In general, response rates are higher in patients with HD or low-grade lymphoma than in those with high-grade lymphoma. Unfortunately, DLI is limited by potentially fatal complications that arise from alloreactive T cells also present in the lymphocyte infusion,8 as the benefit of any GvL effect is often directly associated with graft-versus-host disease (GvHD).
The first reports of successful DLI in lymphoma patients were in those who developed EBV-post-transplant lymphoproliferative disorder (EBVLPD) following allogeneic SCT.9 DLI was ideal in this setting because this malignancy is normally controlled by an EBV-specific T-cell response. Since most EBV-seropositive individuals have a high frequency of EBVspecific precursors, the transfer of unmanipulated DLI should be able to restore the immune response to EBV. When DLI was used in this setting, the overall response rate was high, with 20 of 22 patients achieving complete remissions in one study.10 However, other centers have seen lower response rates to DLI, possibly reflecting differing patient populations or suboptimal EBV-specific immune responses in donors.11
There have also been several groups who have used DLI for the treatment of residual HD or NHL following allogeneic SCT, with better successes reported in low-grade (over 60%) compared with high-grade lymphomas.3,6,7,12 The presence of any significant graft-versus-Hodgkin lymphoma activity following allogeneic SCT has proved difficult to establish, and may be hampered by the high treatment-related mortality rates in this heavily pre-treated patient population. However, transplant registry-based studies report that relapse rates are lower in HD patients who develop GvHD than in those who do not.13,14
Based on this information, Peggs et al. administered DLI to 16 patients with residual disease or disease progression of HD following a reducedintensity conditioning regimen resulting in nine disease responses, including eight complete responses. However, severe, acute GvHD developed in six and chronic GvHD in five.4 More recently, Anderlini et al. reported a response rate of 44% for nine patients with advanced HD who received DLI (median CD3+ cell dose, 77.5×106/kg) for persistent or progressive disease; however, all but one developed GvHD following DLI and no correlation was observed between CD3+ cell dose and disease response.15 Further investigations of DLI approaches in lymphomas are clearly warranted; however, the development of strategies to maximize efficacy and minimize toxicity is crucial if this therapy is to play a major role in the treatment of lymphomas.
Autologously Expanded Non-specific T Cells
Enhanced recovery of lymphocyte counts has been correlated with improved outcome following autologous SCT for both HD and NHL.16 To expand on this concept, one group recently administered anti-CD3 and anti-CD28 ex vivo expanded T cells in a dose-escalation study to 16 patients with refractory or relapsed NHL 14 days after autologous SCT.17 Despite T-cell dysfunction prior to T-cell infusion, all of the infused T cells had normal function and cytokine secretion profiles. Clinical responses included four complete remissions, seven partial remissions, and four with stable disease,17 suggesting that adoptive transfer of autologous CD3/28-stimulated T cells is feasible in heavily pretreated patients with advanced NHL and warrants further study.
Cytokine-induced killer cells (CIK) are a unique population of cytotoxic T lymphocytes (CTL) with phenotypic and functional attributes of T and natural killer (NK) cells. They are a characteristic CD3+ CD56+ phenotype that have been used in the autologous setting to treat relapse or minimal residual disease. In a phase I dose-escalation study, nine patients with relapsed HD or NHL received up to three infusions of autologous CIK, resulting in two partial responses and one stable disease; however, all clinical responses were only transient.18 While it is not clear whether this approach will be efficacious in permitting durable clinical responses of relapsed disease, there may be some benefit in the setting of minimal residual disease.
Cytotoxic T-lymphocyte Therapies
Epstein-Barr Virus-specific Cytotoxic T-lymphocyte Therapies
Viral-associated tumor antigens have an important role as potential immunotherapeutic targets because they may have several unique epitopes that are highly immunogenic targets for a T-cell response. Virus-transformed tumor cells can express an array of viral antigens to be presented for CTL recognition, making these cells more susceptible to T-lymphocyte-mediated immunotherapy than many malignant cells, which may express only small quantities of a single modified peptide as a CTL target. For example, latent EBV infection may be associated with a subset of HD, as well as a variety of NHL, including Burkitt’s lymphoma, NK/T cell lymphoma, and LPD.19–21 Three distinct types of EBV latency with varying expression of EBV latent proteins have been characterized. Tumor cells expressing latency type 1, found in EBVpositive Burkitt’s lymphoma, are not a good target for immunotherapy approaches because EBV nuclear antigen (EBNA)-1, which is the major protein expressed in these tumors, is not well processed by the class I processing machinery.22 In contrast, type 2 latency, which is the hallmark of EBV-positive HD, has a slightly less restricted array of proteins, including EBNA-1 and latent membrane proteins (LMP)-1 and LMP2, the latter two of which are potential T-cell targets. Finally, type 3 latency is seen in EBV-LPD following solid-organ transplantation (SOT) or SCT, and is the ideal model to evaluate the utility of immunotherapy using EBV-specific CTL. This is because the tumor cells are highly immunogenic, expressing all nine latent cycle EBV antigens, including the immunodominant EBNA3 and EBNA2 antigens, as well as the two viral membrane proteins LMP1 and LMP2.
The successful generation of antigen-specific CTL ex vivo requires an antigen-presenting cell (APC), which can effectively present the antigen to T cells as well as a defined antigen. Due to the challenge in generating T cells specific for tumor antigens in which the malignant cell presents antigen poorly and the target antigens are weak, the first studies using antigen-specific CTL focused on targeting type 3 latent EBV antigens, which are expressed in lymphoblastoid cell lines (LCL) that can be readily produced by infecting B cells in vitro with a laboratory strain of EBV.
Post-transplant Lymphoproliferative Disease
Recipients of Stem Cell Transplants
Consequently, our group and others have used EBV-specific CTL (EBV-CTL) generated using donor lymphocytes stimulated with EBV-transformed B cells to expand an EBV-specific CTL population.19,23–26 We have now used donor-derived EBV-CTL as prophylaxis for EBV-induced lymphomas in over 60 patients post-SCT who received an ex vivo T-cell-depleted SCT or those transplanted for an EBV-related malignancy.19,25,27 None of the patients developed EBV-LPD compared with 11.5% in a historical non-treated control group.25 Gene marking confirmed the persistence of these CTL in vivo for as long as seven years post-infusion.28 Similar results were seen by another group, who used EBV-CTL as prophylaxis in six T-cell depleted SCT recipients, with five of the six achieving a reduction in viral load.26 Immunotherapy using EBV-CTL has also been used to treat overt lymphoma, with five of six patients treated successfully.25
Recipients of Solid-organ Transplants
Generating EBV-CTL for clinical use after SOT offers a unique dilemma in that the SOT donor is not human leukocyte antigen (HLA)-matched, and the EBV-associated LPD/lymphoma arises from recipient lymphocytes rather than being donor-derived. Therefore, the vast majority of studies have used autologously generated EBV-CTL.29–31 While many of these studies indicate that autologous EBV-CTL can induce remissions of EBVLPD, the persistence of these CTL appears to be less than that observed using donor-derived CTLs, implying that long-term immunosuppressive therapy in the SOT recipients may compromise CTL persistence and function. Since autologous CTL generation is a time-consuming process, two groups have used banked allogeneic partially HLA-matched EBV-CTL with some success. However, these results were confounded as some patients received additional antiviral therapies and some had dose reductions of their immunosuppressives, leaving the contribution of the allogeneic CTL in question.32,33
Epstein-Barr Virus-associated Lymphomas
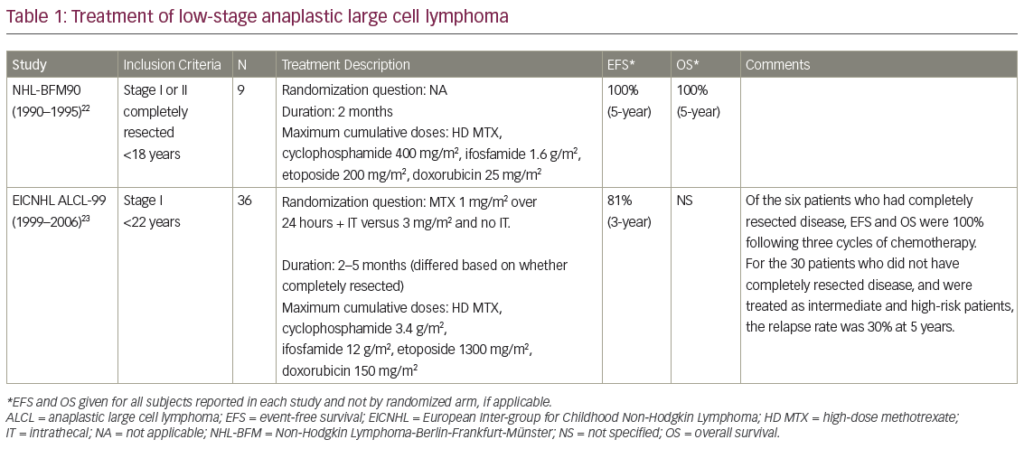
Expressing Type 2 Latency
EBV-positive HD (EBV-HD) and NHL develop in the immunocompetent host where viral gene expression is limited to immunosubdominant proteins, including LMP1 and LMP2, which are weak targets for CTL activity, thereby allowing malignant cells to evade the immune system. Nevertheless, immunotherapy approaches targeting these subdominant EBV antigens have met with some success. In a phase I dose-escalation study, our group evaluated the use of autologous EBV-CTL for 14 patients with relapsed EBV-HD, retrovirally marking CTL in seven of the patients. Clinically, the EBV-CTL were well tolerated and showed antitumor activity, as evidenced by five complete remissions (two of whom had detectable disease at the time of CTL infusion), one partial response, and five with stable disease. Tetramer and functional analyses revealed that T cells reactive with LMP2 were present in the infused lines, expanded in peripheral blood following infusion, and could track to the sites of the disease.34 Gene-marking studies proved that infused effector cells could further expand by several logarithms in vivo with persistence up to 12 months.34
As it is difficult to generate autologous CTL in sufficient enough numbers for heavily pre-treated patients, partially HLA-matched allogeneic CTL were produced for use in a phase I study in patients with relapsed EBV-HD. Five of six patients experienced a reduction in measurable disease with a maximum duration of response of 22 months. However, these patients also received fludarabine conditioning prior to CTL infusion, making it difficult to interpret the role of CTL in the clinical response. Additionally, this approach may be limited by the short-term persistence of the allogeneic cells as donor EBV-CTL could not be detected in vivo.35
Since our previous experience using EBV-CTL for relapsed EBV-HD generated CTL lines with low frequencies of cells specific for LMP2, we have now focused our efforts on using genetically modified tumor APCs that overexpress LMP2 as a strategy to increase the frequency of LMP2-specific EBV-CTL. To do this, we replaced the LCLs as APCs with dendritic cells (DCs) engineered to express LMP2 from an adenovirus (Ad) vector for the primary stimulation, and then used the LCLs overexpressing LMP2 with the same Ad vector for subsequent stimulations (see Figure 1).36 We have now used these genetically engineered LMP2-specific CTL in a dose-escalation study for 16 patients with high-risk EBV-HD and NHL. Ten patients received CTL as adjuvant therapy with nine remaining in complete remission for up to 37 months.37 In addition, five of six patients with active, relapsed disease at the time of LMP2-specific CTL administration showed a tumor response, which was complete in four and sustained for more than nine months. No short- or long-term toxicities were observed after CTL infusion.37 To expand on this concept, we are now extending these studies using autologous T cells enriched for both LMP1 and LMP2 for the treatment of EBV-positive lymphoma, with a clinical trial under way.
Chimeric T-cell Therapies
Following CTL therapy, subpopulations of EBV-positive HD tumor cells may lack or lose expression of the weak immunogenic antigens (LMP1, LMP2, and EBNA-1), allowing tumor escape and subsequent treatment failure. To overcome this dilemma, our group has generated EBV-CTL grafted with a chimeric antigen receptor (CAR) targeting the CD30 molecule, which is overexpressed on malignant Hodgkin Reed-Sternberg cells found in HD tumors.29 We have shown that these CD30CAR+ EBVCTL retain their ability to kill EBV-positive tumors and acquire the ability to recognize and kill CD30-positive HD tumor cells in vitro and in a severe combined immuno-deficiency (SCID) mouse model in vivo. This work is now being translated into the clinical setting, and may enhance immunotherapy of patients with EBV-positive and EBV-negative HD.29
Strategies to Improve Ex Vivo Generation of T Cells
Perhaps the greatest limitation of CTL therapy is the time needed for ex vivo expansion. The rate-limiting step in the generation of these CTL is the time necessary to generate sufficient numbers of APCs that are used to generate antigen-specific CTL. A number of approaches using artificial antigen-specific cells, such as grafting ligands for the T-cell receptor and co-stimulatory surface molecules onto mice fibroblasts or beads, have been evaluated.38–40 Another limitation is the lack of in vivo persistence of infused cells. Several groups have tried to identify the optimal phenotype of infused T cells, which likely contains CD4 and CD8 effector and memory cells. Meanwhile, it is imperative to correlate in vivo function with the source of antigen-presenting cells and T-cell culture conditions.
Depletion of Regulatory T Cells
Recently, focus has turned to a subpopulation of CD4 cells known as regulatory T cells, which co-express intracellular Forkhead box (FoxP)3 and the alpha chain of the IL-2 receptor, CD25. These cells are crucial in controlling the response to foreign antigens, allowing protection against autoimmune diseases.41 Increasing evidence also suggests a role for these cells in offering an immune escape mechanism in HD, by suppressing antitumor immune responses. In HD tumors, a substantial percentage of the T-cell infiltrate consists of Tregs,42 and the number of these cells, marked by expression of FoxP3, is associated with survival in HD patients. Tregs have been shown to be elevated in the peripheral blood of patients with newly diagnosed and relapsed HD compared with healthy controls.43 It is also known that Tregs suppress interferon-gamma production by lymphocytes, including the EBV-specific CD8+ T-cells. For this reason, several groups have evaluated strategies to deplete this immunesuppressive population prior to the administration of tumor-specific cytotoxic T cells.
Lymphodepletion Strategies
Methods allowing for depletion of Tregs, such as selective depletion of CD25 or the use of T- and B-cell-depleting antibodies such as alemtuzumab, could be beneficial in the lymphoma setting. Several groups have also evaluated whether administering lymphodepleting chemotherapy, such as fludarabine or monoclonal antibodies, may not only deplete Tregs but also encourage a better environment to promote homeostatic expansion of infused T cells.44,45 Clinical studies had already shown expansion of T cells specifically for antigens expressed on solid tumors such as melanoma following lymphodepletion.46 In the lymphoma setting, Peggs et al. recently reported results of two phase II national prospective studies of matched sibling donor SCTs in 67 patients with multiple-relapsed HD comparing reduced-intensity conditioning consisting of fludarabine and melphalan with differing GvHD prophylaxis.5 One group received alemtuzumab (anti-CD52) with cyclosporine and the other short-course methotrexate with cyclosporine. Progression-free survival was superior in the group receiving alemtuzumab (four-year progression-free survival (PFS) 39 versus 25%), with significantly lower incidences of non-relapse mortality and GvHD; the authors attributed these results to durable responses to DLI following inclusion of alemtuzumab in the conditioning protocol.5
Escaping Immune Evasion Mechanisms
Immune escape by tumor cells is made possible by a variety of mechanisms, such as the secretion of inhibitory cytokines by the malignant cells or downregulation of major histocompatibility complex and co-stimulatory molecules. For example, transforming growth factor- β (TGF-β), which is secreted by a wide variety of tumors, has detrimental effects on CTL proliferation and function.47,48 In fact, transgenic mice genetically engineered so that their T cells are insensitive to TGF signaling are able to eradicate tumors.49
Our group has been interested in the role of dominant-negative TGF-β type 2 receptor (DNR)-transduced CTL in eradicating tumors given that others have shown that the adoptive transfer of such cells is able to eradicate tumor cells in murine models.50 We have shown in human in vitro studies that DNR-transduced CTL were resistant to the antiproliferative effects of recombinant TGF-β and long-term expression of this construct had no deleterious effects on the function, phenotype, or growth characteristics of the transduced lines.51 Thus, CTL expressing the DNR may have a selective advantage in vivo in TGF-β secreting tumors. We will shortly open a phase I trial to test this hypothesis now that murine safety studies have been completed.52
Conclusions
Adoptive immunotherapy offers an avenue of potentially curative therapy for patients with multiple relapsed or refractory lymphomas, ranging from the simple transfer of unmanipulated donor T cells to time-consuming and complex CTL therapies. As researchers optimize the generation of these cells ex vivo, allowing for enhanced in vivo persistence and expansion, we will begin to see more durable responses in this heavily pre-treated population.