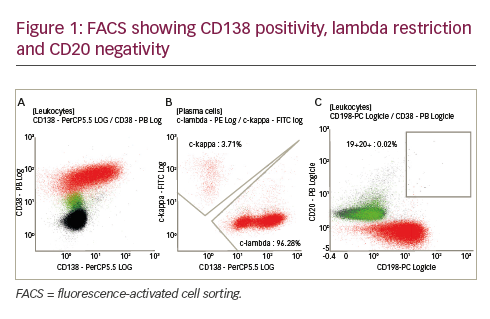
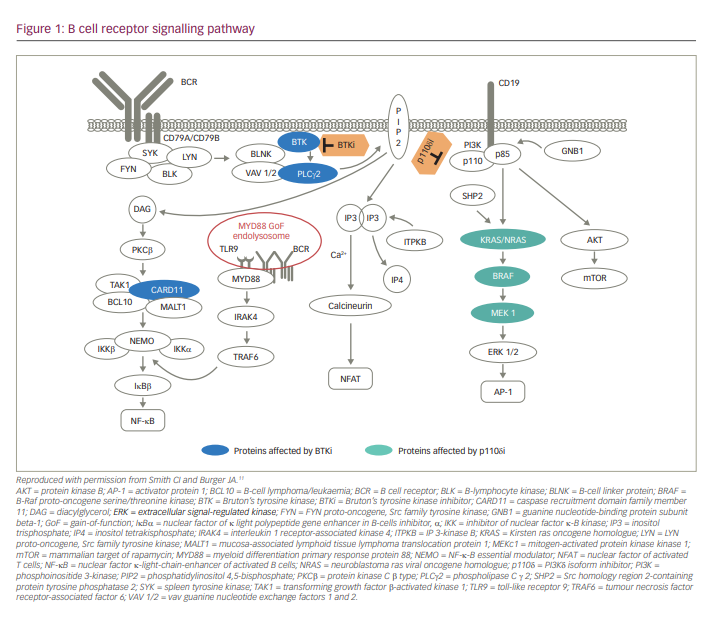
B-cell signalling pathways are critical regulators of B-cell development, expansion, and survival and function via various mediators, including Src family kinases (SFKs), spleen tyrosine kinase, Bruton’s tyrosine kinase (BTK) and phosphatidylinositol 3 kinase (PI3K) (Figure 1).1,2 BTK, a member of the Tec protein tyrosine kinase (TEC) family, is overexpressed in many B-cell leukaemias and lymphomas, and regulates survival, proliferation, adhesion and migration of malignant B cells.3–6 Dysregulated BTK signalling has been implicated in the pathogenesis of chronic lymphocytic leukaemia (CLL),3 mantle cell lymphoma (MCL),7 diffuse large B-cell lymphoma (DLBCL),5,8 Waldenström’s macroglobulinaemia (WM)9 and marginal zone lymphoma (MZL).6 Currently, selective inhibition of BTK is an important strategy for the chemotherapy-free management of B-cell malignancies.10
Ibrutinib therapy for B-cell malignancies: promises and challenges
BTK is a clinically validated target in the treatment of B-cell malignancies, as demonstrated by the efficacy of the first-generation BTK inhibitor (BTKi), ibrutinib.5 Ibrutinib (PCI-32765) is a first-in-class, oral, irreversible BTKi that binds cysteine 481 in the kinase domain of BTK to block its activity, allowing for chemotherapy-free treatment. Ibrutinib received approval for relapsed/refractory (R/R) MCL in 2013, for R/R CLL/small lymphocytic lymphoma (SLL) in 2014, for WM in 2015 and for R/R MZL in 2017.10,12 In treatment-naïve patients with CLL/SLL, ibrutinib therapy was approved in 2016, and ibrutinib/rituximab therapy was approved in 2020.10
Although ibrutinib therapy is generally well tolerated and has a significant clinical benefit over chemotherapy in CLL/SLL,13,14 it has a toxicity profile that is attributed to off-target inhibition of kinases structurally related to BTK, including epidermal growth factor receptor (EGFR), TEC, Janus kinase 3 (JAK3) and SFKs.15–18 Ibrutinib treatment is associated with a significant risk of atrial fibrillation (AF; cumulative incidence rate: 11.2%) and heart failure (3-year risk: 7.7%), possibly due to the off-target inhibition of PI3K-AKT signalling.19–23 Ibrutinib treatment also has a significant risk of late-onset hypertension, which is associated with a long-term risk of developing other major cardiac events.20,24 The BTK–TEC pathway mediates platelet function, and ibrutinib use was shown to be associated with an increased risk of bleeding, especially in older patients and in those receiving antithrombotic treatment.22,25–27 Gastrointestinal and skin toxicities associated with ibrutinib therapy may be the result of the off-target inhibition of EGFR signalling.28–31 Ibrutinib intolerance and disease progression are the main factors dictating treatment discontinuation.32–35 BTKis with a better selectivity profile could benefit patients who become intolerant to ibrutinib.36
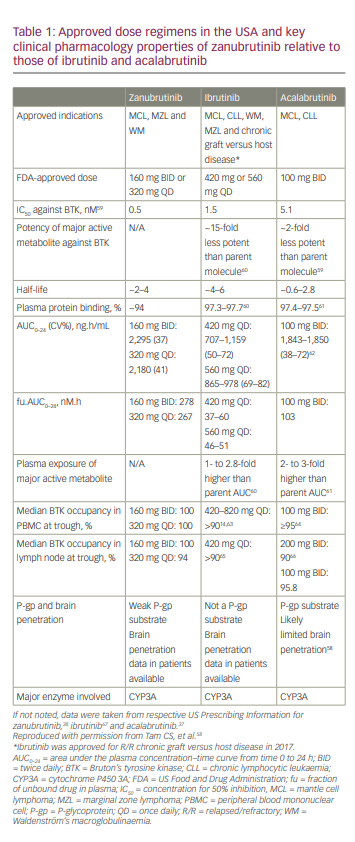
Acalabrutinib and zanubrutinib (first approved in 2017 and 2019, respectively) are next-generation BTKis that were developed to reduce the off-target effects associated with ibrutinib.10,18,37,38 Acalabrutinib (ACP 196) is an irreversible BTKi that was rationally designed to have higher target selectivity than ibrutinib.17,39 The efficacy and safety of acalabrutinib were shown in trials for R/R MCL, CLL and WM.17,39–41 Zanubrutinib (BGB-3111) is a highly potent and selective next-generation BTKi designed to provide complete and sustained BTK occupancy for efficacy across disease-relevant tissues, with theoretically fewer off-target adverse events compared with ibrutinib.18,42 Other orally available BTKis currently in clinical development include the following: 1) orelabrutinib, a highly selective irreversible BTKi that showed efficacy in R/R CLL, MCL and R/R WM;43,44 2) pirtobrutinib (LOXO-305), a highly selective, third-generation, reversible BTKi that showed efficacy in heavily pretreated patients with CLL, MCL or WM who were intolerant/resistant to previous BTKi treatment;32 and 3) nemtabrutinib (MK-1026 or ARQ-531), a non-covalent, reversible BTKi undergoing late-stage clinical development for B-cell malignancies.45
Zanubrutinib: United States Food and Drug Administration approval and mechanism of action
Based on data from two key phase II studies in MCL46 and MZL,47 zanubrutinib received accelerated approval by the United States Food and Drug Administration in 2019 for the treatment of MCL in adults who have received ≥1 prior therapy, and in 2021 for patients with R/R MZL who have received ≥1 anti- CD20-based regimen. Other studies evaluating the efficacy and safety of zanubrutinib include a phase II study in R/R MZL,48 a phase III study in WM,16 a phase II study in DLBCL,49 a phase III study in R/R CLL/SLL,50 a phase III study in treatment-naïve patients with CLL/SLL with and without del(17p)51 and phase II study in patients with previously treated B-cell lymphoma that was ibrutinib and/or acalabrutinib intolerant.52 Zanubrutinib was approved for WM in the USA and EU in 2021;38,53 for R/R CLL and R/R MCL in 2020 and R/R WM in 2021 in China;54–56 for R/R MCL and WM in Canada in 2021;57 for R/R WM in the UK in 2021; for R/R MCL and R/R WM in Australia in 2021; for R/R WM in Estonia, Iceland, Ireland, Netherlands and Norway in 2021; for R/R MCL in Ecuador, Chile, United Arab Emirates, Saudi Arabia and Israel in 2021; and for R/R MCL and R/R WM in Taiwan in 2021. Zanubrutinib has a molecular weight of 471.55 Da. The molecular structure is depicted in Figure 2.
Zanubrutinib covalently binds with high specificity to the BTK adenosine triphosphate binding pocket at cysteine 481 to deliver irreversible, sustained kinase inhibition.18,42 In a phase I study of patients with B-cell malignancies, median BTK occupancy in peripheral blood mononuclear cells (PBMCs) was >95% at all doses tested; in lymph-node tissue, a higher proportion of patients achieved >95% BTK occupancy at trough with the 160 mg twice daily (BID) dosing regimen compared with the 320 mg once daily (QD) regimen (89% versus 50%, respectively).48 The high, sustained BTK inhibition in both PBMCs and lymph nodes maximizes the chances for deep and sustained remission.18,48,50
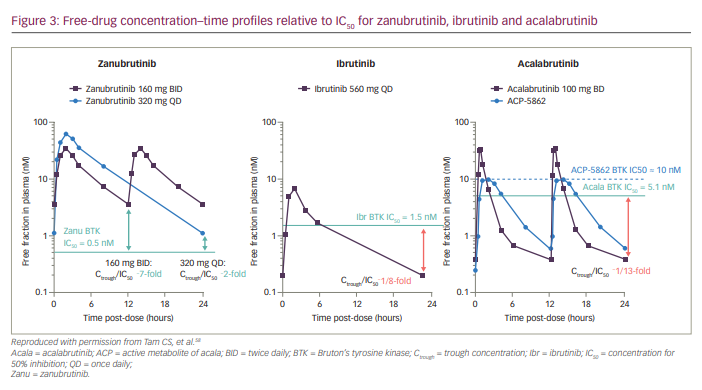
Compared with ibrutinib, zanubrutinib binds with minimal affinity to EGFR (3.5× less), JAK3 (58× less), human EGFR2 (28× less), interleukin (IL)-2-inducible T-cell kinase (19× less) and TEC protein tyrosine kinase (3.5× less).18 Zanubrutinib has similar or increased potency compared with other BTKis (concentration for 50% inhibition [IC50] values for zanubrutinib, ibrutinib and acalabrutinib are 0.5 nM, 1.5 nM and 5.1 nM, respectively).58 The unbound fraction of zanubrutinib in plasma is approximately twice that of ibrutinib or acalabrutinib (~6% versus ~3%; Table 1).58
Pharmacokinetics and pharmacodynamics of zanubrutinib
Based on the pharmacokinetic (PK) profile of zanubrutinib after its single- and multiple-dose administration in patients with B-cell malignancies (ClinicalTrials identifier: NCT02343120),48 the recommended dosing of zanubrutinib is 160 mg orally BID or 320 mg orally QD.48,58 Orally administered zanubrutinib was rapidly absorbed (peak plasma concentration [Cmax] 2 hours after dosing). The mean terminal elimination half-life (t1/2) was ~2–4 hours; the median time to peak plasma concentration (Tmax) was ~2 hours. Systemic accumulation was limited after repeated dosing.58 The Ctrough/IC50 ratios were 7 and 2 for zanubrutinib doses at 160 mg BID and 320 mg QD, respectively. Therefore, zanubrutinib plasma concentration was consistently above the IC50 during the 24-hour dosing schedule for both dosing regimens. The Ctrough/IC50 ratio for zanubrutinib was higher than that of ibrutinib and acalabrutinib (Figure 3).58
Zanubrutinib can be administered with or without food,58 and there were no clinically significant differences in area under the curve (AUC) or Cmax following a high-fat meal in healthy subjects (ClinicalTrials.gov identifier: NCT04163523).38,58 The absorption of zanubrutinib was not affected by gastric-acid-reducing agents, and it can be taken concomitantly with proton-pump inhibitors and acid-reducing agents without dose adjustment.58 The PK of zanubrutinib was not significantly affected by race (Asian versus non-Asian)68 or by mild/moderate hepatic impairment.69
Zanubrutinib undergoes extensive metabolism via a cytochrome P450 3A (CYP3A) pathway and has no active metabolites in circulation; in contrast, ibrutinib and acalabrutinib both have major active metabolites in circulation.58,68,70 Co-administration with moderate or strong CYP3A inhibitors (e.g. azole antifungals) increased zanubrutinib AUC and Cmax, thereby increasing the risk of zanubrutinib toxicity. Co-administration with CYP3A inducers (e.g. rifampin) decreased zanubrutinib AUC and Cmax. Dose reduction is therefore recommended when zanubrutinib is administered with moderate-to-strong CYP3A inhibitors, but not with weak CYP3A inhibitors/inducers.70
The pharmacodynamic assessment of zanubrutinib-mediated BTK inhibition in pre- and on-treatment lymph nodes and in PBMCs showed complete and sustained BTK inhibition at 160 mg BID (100% median steady-state BTK occupancy in both lymph nodes and PBMCs) and at 320 mg QD (94% median steady-state BTK occupancy in lymph nodes and 100% occupancy in PBMCs; Table 1).48 BTK occupancy was comparable or higher than that for acalabrutinib and ibrutinib.58 Zanubrutinib was also shown to have no clinically relevant effects on cardiac repolarization at therapeutic as well as supratherapeutic doses.71,72
Based on the low susceptibility of zanubrutinib to PK modulation by intrinsic and extrinsic factors (e.g. hepatic impairment, food effects) leading to consistent, sustained therapeutic exposures, and its ability to be prescribed concomitantly with proton-pump inhibitors and moderate/strong CYP3A4 inhibitors (with reduced dose), zanubrutinib is considered a safe and effective treatment option when prescribed according to its approved label.
Evaluation of the efficacy and safety of zanubrutinib
Key studies evaluating the efficacy and safety of zanubrutinib include the following: ASPEN, a randomized, open-label, phase III study, which was the first head-to-head comparison of zanubrutinib versus ibrutinib in patients with myeloid differentiation primary response 88 (MYD88)-mutant WM and in patients with wild-type MYD88;16,73 ALPINE, an on-going, head-to-head, phase III, randomized, open-label study comparing the efficacy and safety of zanubrutinib versus ibrutinib in patients with R/R CLL/SLL;50 and SEQUOIA, an on-going, global, open-label, randomized, phase III study comparing the efficacy of zanubrutinib versus bendamustine plus rituximab (BR) in treatment-naïve patients with CLL/SLL with or without del(17p).51
The ASPEN trial recruited patients with MYD88L265P WM who were either R/R (>1 prior line of treatment) or treatment naïve and unsuitable for standard immunochemotherapy. Prior BTKi treatment was exclusionary.16 Patients in Cohort 1 were randomized to receive either 160 mg zanubrutinib BID (Arm A, n=102) or 420 mg ibrutinib QD (Arm B, n=99). In Cohort 2, patients with MYD88WT WM received 160 mg zanubrutinib BID (Arm C). The primary endpoint was the proportion of patients achieving a complete response (CR) or very good partial response (VGPR) by independent review. Key secondary endpoints included major response rate, progression-free survival (PFS), duration of response (DOR), disease burden and safety.16 At a median follow-up of 19.4 months, no patient achieved CR. Although the study did not meet its statistical primary endpoint for superiority, there was a trend towards better disease control for zanubrutinib versus ibrutinib; independent review committee (IRC)-assessed VGPR was higher in the zanubrutinib arm compared with the ibrutinib arm (28% versus 19%, respectively, two-sided p=0.09). PFS rates at 18 months were 84% with ibrutinib and 85% with zanubrutinib, while estimated overall survival (OS) rates at 18 months were 93% with ibrutinib and 97% with zanubrutinib.16
The ASPEN study reported that neutropenia, upper respiratory tract infections and diarrhoea were the most common all-grade AEs among zanubrutinib-treated patients (n=101), whereas diarrhoea, upper respiratory tract infections, contusion and muscle spasms were the most common all-grade AEs among ibrutinib-treated patients (n=98).16 Ibrutinib-treated patients had a ≥10% higher incidence of AF, diarrhoea, contusion, muscle spasms, peripheral oedema and pneumonia compared with zanubrutinib-treated patients, whereas the incidence of neutropenia was ≥10% higher among zanubrutinib-treated patients (grade ≥3 neutropenia was ≥5% higher). Nonetheless, grade ≥3 infection rates were comparable in both arms (1.2 and 1.1 events per 100 person-months for ibrutinib and zanubrutinib, respectively).16 Ibrutinib-treated patients had a ≥5% higher incidence of grade ≥3 hypertension and pneumonia compared with zanubrutinib-treated patients. More patients treated with ibrutinib than zanubrutinib required dose reductions due to AEs (23% versus 14%, respectively).16 The zanubrutinib group showed a greater improvement versus ibrutinib in most quality-of-life measures.16 A sub-study (Cohort 2) of the ASPEN trial reported that zanubrutinib also induced high-quality responses in patients with MYD88WT WM; 27% of MYD88WT patients had VGPRs or better, but there were no CRs. Although cross-study comparisons are not reliable, these rates were comparable to rates in zanubrutinib-treated patients with R/R and treatment-naïve MYD88MUT disease and identical to VGPRs in patients with MYD88WT disease treated with ibrutinib plus rituximab.16,74,75 The safety profiles were similar in zanubrutinib-treated MYD88WT patients and those with MYD88MUT disease.75
The on-going ALPINE trial, which is currently the only comparative BTKi study that includes zanubrutinib, compares the efficacy and safety of zanubrutinib versus ibrutinib in ~600 patients with R/R CLL/SLL who have received one or more prior systemic therapies.50 Prior BTKi therapy is exclusionary. The primary endpoint of the ALPINE study is the overall response rate (ORR: partial response [PR] + CR) by investigator assessment. The first interim analysis included 207 patients who were randomized to zanubrutinib (Arm A: 160 mg BID) and 208 patients who were randomized to ibrutinib (Arm B: 420 mg QD). Median follow-up was 15.3 months for Arm A and 15.4 months for Arm B. ORR was significantly higher in Arm A than in Arm B (78.3% versus 62.5%, respectively; p=0.0006).76 Arm A had higher ORR than Arm B in patients with del(17p) (83% versus 54%, respectively) and in key subgroups including age (<65 years versus ≥65 years, respectively), sex, disease stage, number of lines of prior therapy (1–3 versus >3, respectively), baseline del(17p)/TP53 mutation status (80% for Arm A versus 50% for Arm B) and bulky disease (80% for Arm A versus 64% for Arm B).76 Arm A had higher 12-month PFS rates (94.9% versus 84.0%; hazard ratio [HR] 0.40; 95% confidence interval [CI] 0.23–0.69; p=0.0007) and a trend towards higher 12-month OS rates (97.0% versus 92.7%; HR 0.54; 95% CI 0.25–1.16; p=0.1081) compared with Arm B.76
At the first interim analysis, the ALPINE study reported that 55.9% of patients in Arm A had grade ≥3 AEs compared with 51.2% in Arm B. AEs leading to treatment discontinuation occurred in 7.8% and 13% of patients in Arm A and Arm B, respectively.76 Neutropenia (any grade) occurred in 28.4% and 21.7% of Arm A and Arm B, respectively, whereas grade ≥3 neutropenia was seen in 18.6% and 15.0% of patients, respectively. The higher rate of neutropenia did not translate into a higher rate of infections; any-grade infections occurred in 59.8% and 63.3% of patients, respectively, and grade ≥3 infections occurred in 12.7% and 17.9% of patients, respectively.76 The rates of hypertension were similar in the two treatment arms (16.7% versus 16.4% for any grade, and 10.8% versus 10.6% for grade ≥3, respectively).76 Major haemorrhage (any grade) occurred in 2.9% and 3.9% of patients in the two arms, respectively.76 The rate of AF and atrial flutter (any grade) was significantly lower in Arm A compared with Arm B (2.5% versus 10.1%, respectively, two-sided p=0.0014). AF and flutter (grade ≥3) occurred in 1% and 1.9% of patients in the two arms, respectively. Treatment was discontinued due to cardiac disorders in seven patients in the ibrutinib arm.76 The superior response rate, improved PFS and lower rate of AF/flutter compared with ibrutinib supported the potential use of zanubrutinib for R/R CLL/SLL. The ALPINE study limitations include the relatively short follow-up time to date, the use of lymphoma response criteria rather than the revised criteria including PR-L, and the unblinded nature of the study with investigator-assessed ORR primary endpoint.76
The on-going SEQUOIA trial was designed to compare the efficacy of zanubrutinib versus a combination of BR in treatment-naïve patients with CLL/SLL. Patients with CLL and del(17p) mutation show marked resistance to genotoxic chemotherapies, which cannot be overcome by incorporating anti-CD20 antibodies.77 Additionally, most patients with del(17p) lack wild-type TP53, leading to genomic instability and poor responsiveness to chemotherapy.77 Therefore, Cohort 1 included participants without del(17p), whereas participants with del(17p) were enrolled in Cohorts 2 and 3.51 Participants in Cohort 1 were randomized 1:1 to zanubrutinib (Arm A, n=241) or six cycles of BR (Arm B, n=238). Randomization was stratified by age, Binet stage, immunoglobulin variable region heavy chain (IGHV) mutational status and geographical region. The primary endpoint for Cohort 1 was PFS by IRC, and key secondary endpoints were PFS by investigator assessment, ORR (per IRC and investigator assessment), OS and safety.78 The median age was 70 years in both arms; 29.0% and 29.4% of patients in the zanubrutinib and BR arms had Binet Stage C disease, respectively; unmutated IGHV occurred in 53.4% of patients in the zanubrutinib arm versus 52.4% of patients in the BR arm.
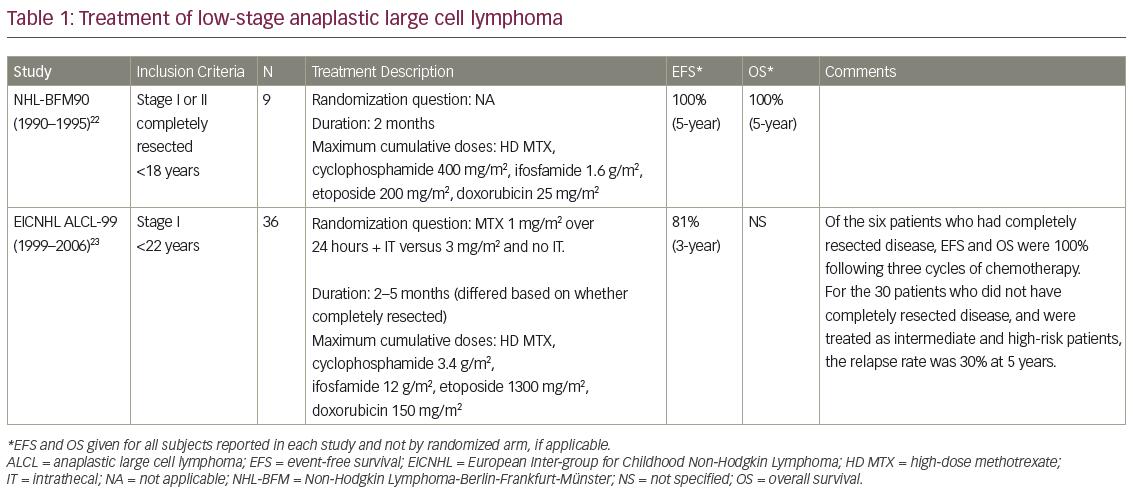
The 24-month PFS rates per IRC assessment were 85.5% and 69.5% in the zanubrutinib and BR arms, respectively (HR 0.42; 95% CI 0.27–0.63; p<0.0001).78 PFS rates by IRC were also higher in the zanubrutinib arm compared with the BR arm in key subgroups, including age (<65 versus ≥65 years, respectively), sex, Binet stage (A or B versus C, respectively), bulky disease (<5 cm versus ≥5 cm, respectively), cytopenia at baseline, chromosome 11q deletion and IGHV mutational status (mutated versus unmutated).78
The SEQUOIA study reported 11 deaths each in the zanubrutinib and the BR arms at a median follow-up of 26.4 and 25.9 months, respectively.78 Grade ≥3 AEs were recorded in 52.5% and 79.7% of patients in the zanubrutinib and BR arms, respectively.78 AEs leading to dose reduction were recorded in 7.5% and 37.4% of patients in the zanubrutinib and BR arms, respectively. AEs leading to dose interruption or delay were recorded in 46.3% and 67.8% of patients in the two arms, respectively, and AEs leading to discontinuation of treatment were recorded in 8.3% and 13.7% of patients in the two arms, respectively.78 In the zanubrutinib and BR arms, 11.7% and 51.1% of patients had grade ≥3 neutropenia; 2.1% and 7.9% had grade ≥3 thrombocytopenia; 0.4% and 1.3% had grade ≥3 AF; 3.8% and 1.8% had grade ≥3 major bleeding; and 6.3% and 4.8% had grade ≥3 hypertension, respectively.78
Cohort 2 of the SEQUOIA trial included 110 patients with del(17p) who received zanubrutinib (160 mg BID; Arm C).51,78 With a median follow-up of 30.5 months, the 24-month PFS was 88.9%.78 Data from Cohorts 1 and 2 of the SEQUOIA trial showed that zanubrutinib was well tolerated, was associated with low AF rates and demonstrated superiority in PFS over the BR regimen per IRC assessment, including in high-risk subgroups (e.g. unmutated IGHV, del[11q]).
Cohort 3 of the SEQUOIA trial included patients with a del(17p)/TP53 mutation, who received zanubrutinib plus venetoclax (Arm D).51,78 The key endpoints included safety, ORR, PFS, DOR and undetectable minimal residual disease (uMRD).79 The del(17p)/TP53 mutation safety population included 49 patients who received zanubrutinib (160 mg BID for ~≥27 cycles). Tumour lysis syndrome (TLS) was assessed between baseline and the end of cycle 3 of zanubrutinib. Venetoclax ramp-up was initiated after the 3-cycle lead-in of zanubrutinib; then, venetoclax (400 mg QD) was administered for 12–24 cycles.79 The post-baseline efficacy assessment included 36 patients, and the combination treatment included 34 patients.79 The median age of the study population was 65 years; 93.9% and 91.9% of patients were positive for del(17p) and TP53 mutation, respectively, and IGHV was unmutated in 87.8% of patients.79 There were no reports of clinical TLS, and the 3-cycle zanubrutinib lead-in decreased the risk of TLS.79 With a median follow-up of 12 months, 36% of patients who received combination therapy achieved CR/CRi (CR with incomplete bone marrow recovery). Although four additional patients met the criteria for CR/CRi, bone marrow assessment was not performed to confirm CR/CRi.79 No AEs led to dose reduction, 29.4% of AEs led to dose interruption and no AEs led to discontinuation. There were no fatal AEs.79 Data from Cohort 3 (Arm D) of the SEQUOIA trial showed that zanubrutinib plus venetoclax combination treatment resulted in a high response rate in the high-risk del(17p)/TP53 mutation subgroup of patients with CLL/SLL. Responses were deeper with longer durations of treatment, as evidenced by the achievement of CR/CRi and uMRD. The combination was well tolerated.79
Zanubrutinib safety, tolerability and adverse event management
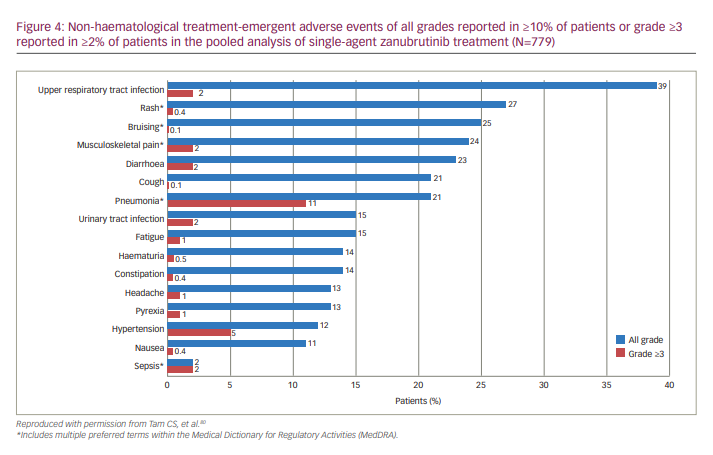
A pooled safety analysis of six studies (N=779 patients with B cell malignancies treated with zanubrutinib) was conducted to understand treatment-emergent AEs and treatment-limiting toxicities of zanubrutinib monotherapy.80 The studies included A Phase I/II, Open-Label, Multiple-Dose, Dose Escalation and Expansion Study to Investigate the Safety and Pharmacokinetics of the BTK Inhibitor BGB-3111 in Subjects With B-Cell Lymphoid Malignancies (ClinicalTrials.gov identifier: NCT02343120; B-cell malignancies, n=385);48 A Phase I Clinical Study to Investigate the Safety, Tolerability, and Pharmacokinetics/Pharmacodynamics of BTK Inhibitor BGB-3111 in Chinese Patients With B-cell Lymphoma (ClinicalTrials.gov identifier: NCT03189524; B-cell lymphomas, n=44);81 A Single-Arm, Open-Label, Multicenter Phase 2 Study to Evaluate Safety and Efficacy of BGB-3111, a Bruton’s Tyrosine Kinase (BTK) Inhibitor in Relapsed or Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma (CLL/SLL) (ClinicalTrials.gov identifier: NCT03206918; CLL/SLL, n=91);82 A Single-Arm, Open-Label, Multicenter Phase 2 Study to Evaluate Efficacy and Safety of BGB-3111, a Bruton’s Tyrosine Kinase (BTK) Inhibitor, in Subjects With Relapsed or Refractory Mantle Cell Lymphoma (MCL) (ClinicalTrials.gov identifier: NCT03206970; MCL, n=86);46 A Phase 2, Single-Arm, Open-Label, Multicenter Study of Bruton’s Tyrosine Kinase (BTK) Inhibitor BGB-3111 in Chinese Subjects With Relapsed/Refractory Waldenström’s Macroglobulinemia (WM) (ClinicalTrials.gov identifier: NCT03332173; WM, n=44);83 and A Phase 3, Randomized, Open-Label, Multicenter Study Comparing the Efficacy and Safety of the Bruton’s Tyrosine Kinase (BTK) Inhibitors BGB-3111 and Ibrutinib in Subjects With Waldenström’s Macroglobulinemia (WM) (ClinicalTrials.gov identifier: NCT03053440; WM, n=129).73 The most common non-haematological toxicities reported were upper respiratory tract infection (39%), rash (27%), bruising (25%), musculoskeletal pain (24%), diarrhoea (23%), cough/pneumonia (21% each), urinary tract infection /fatigue (15% each), haematuria/constipation (14% each), headache/pyrexia (13% each), hypertension (12%) and nausea (11%).80 Grade ≥3 non-haematological treatment-emergent AEs included pneumonia (11%), hypertension (5%), upper respiratory tract infection, urinary tract infection, sepsis, diarrhoea and musculoskeletal pain (2% each) (Figure 4). Bleeding or bruising events were reported in 55% of patients, although most were grade 1 or 2.80 Zanubrutinib had consistently low rates of AEs of interest commonly associated with the BTKi class.80
Additionally, in zanubrutinib-treated patients with R/R B-cell malignancies who were intolerant to prior BTKis, 70% of ibrutinib intolerance events and 83% of acalabrutinib intolerance events did not recur on zanubrutinib, and no events occurred at a higher severity.84 These data support the use of zanubrutinib after intolerance to other BTKis.
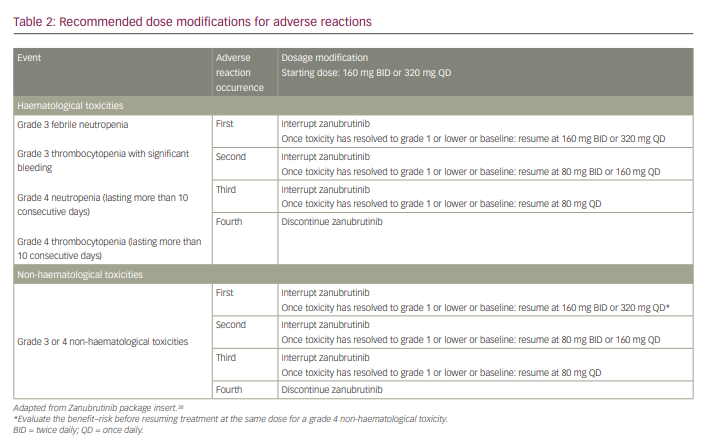
Dosage modifications are recommended for AEs based on recommendations from the US prescribing information (Table 2).38 Dose interruption is recommended after the first, second and third occurrences of any of the following: grade 3 febrile neutropenia, grade 3 thrombocytopenia with significant bleeding, grade 4 neutropenia (lasting >10 consecutive days) and grade 4 thrombocytopenia (lasting >10 consecutive days). Zanubrutinib should be discontinued upon the fourth occurrence of these events and after the fourth occurrence of grade 3 or higher non-haematological toxicities.
Precautions recommended for zanubrutinib therapy include the following: 1) discontinuation of treatment upon any signs of intracranial bleeding and for 3–7 days pre- and post-surgery if there is a risk of bleeding; 2) dosage modification or termination in the case of grade 3 or 4 cytopenias; 3) careful monitoring and appropriate therapy for grade 3 or higher infections (most commonly pneumonia), which occur in more than a quarter of patients treated with zanubrutinib; 4) use of sun protection and regular monitoring for second primary malignancies, as non-melanoma skin cancer was the most common non-primary malignancy in zanubrutinib-treated patients; 5) monitoring for AF and atrial flutter. Patients who take zanubrutinib while pregnant should be informed about potential embryo-fetal toxicity.38
Impact of zanubrutinib on future clinical practice for B-cell malignancies
The emergence of therapeutic modalities such as BTKis, B-cell lymphoma 2 (BCL2)-based therapies, PI3K inhibitor-based therapies and chimeric antigen receptor (CAR) T-cell therapies has altered the treatment landscape for B-cell malignancies. Selection of an appropriate therapeutic regimen is a decision that is shared between the clinician and patient and is dependent on various factors, including 1) patient characteristics (age, fitness, comorbidities and concomitant medications), 2) disease characteristics (presence of prognostic markers, disease bulk, area of involvement, risk of TLS), and 3) regimen characteristics (cost, toxicity and convenience) (Figure 5a). Other parameters include genetic risk and response to prior lines of treatment.77
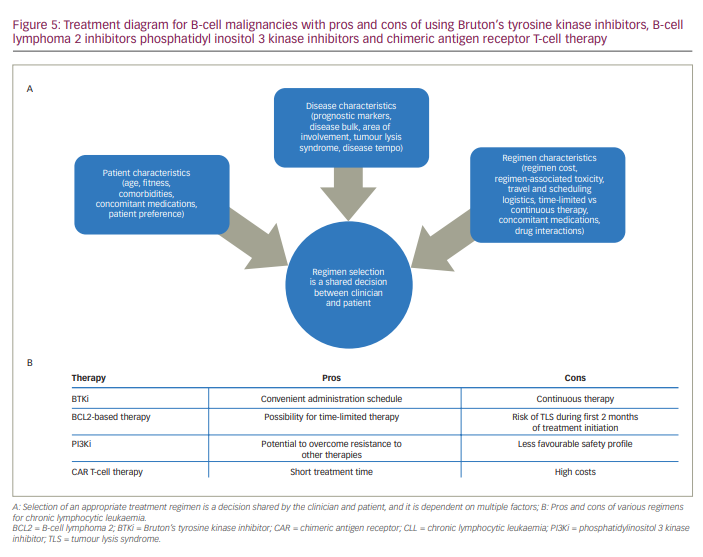
Although targeted therapies such as the BTKis are rapidly replacing chemoimmunotherapeutic regimens for CLL, head-to-head comparisons are lacking among the more recently developed strategies such as BTKis, BCL2-based therapies, PI3Ki-based therapies and CAR T-cell therapies. Some of the advantages and disadvantages of selected therapeutic regimens for CLL are listed in Figure 5b. BTKis require continuous treatment, which can be associated with financial burden, drug toxicity and development of resistance. In contrast, BCL2-based therapies have been developed as 1-year or 2-year fixed-term regimens. Although the time-limited strategy of BCL2-based therapies such as venetoclax may not be suitable for subgroups of patients with specific genetic abnormalities, BCL2-based therapies are associated with the achievement of uMRD.85 PI3Kis such as idelalisib and umbralisib have demonstrated activity in ibrutinib-resistant patients but are often associated with serious toxicities and are only recommended for patients who progress on prior lines of therapy.86 Although CAR T-cell therapy shows promise in R/R CLL, the long-term durability of response is not clear, and logistical considerations may impact therapeutic decisions.87
Conclusions
Selective inhibition of BTK activity is an important modality in targeted precision therapy to treat B-cell malignancies. The first-generation BTKi, ibrutinib, has well-described off-target effects that contribute to its toxicity profile, notably an increased risk for cardiovascular toxicities, including AF, hypertension and haemorrhage. Zanubrutinib is a potent and highly selective next-generation BTKi designed to provide complete and sustained BTK occupancy for efficacy across disease-relevant tissues, with lower rates of certain off-target AEs compared with ibrutinib. Its safety features include minimal cardiovascular toxicity.16 The development of new classes of therapeutic agents (e.g. BTKis, BCL2-based agents, PI3Kis and CAR T-cell therapy) makes it important to study and optimize sequencing strategies carefully and to determine the potential benefits of combination therapies (e.g. BTKi plus venetoclax) in selected high-risk patients.88