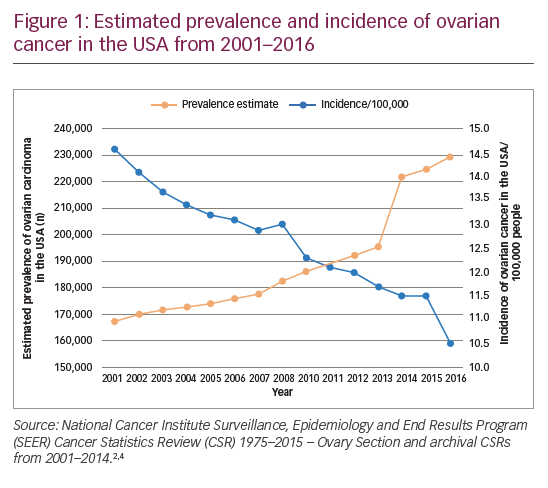
Worldwide, ovarian cancer is the seventh most common cancer and the eighth most common cause of cancer death in women.1 The GLOBOCAN study estimated there were 239,000 new cases in 2012 and 152,000 deaths due to this disease. There are nearly 600,000 women living within 5 years of an ovarian cancer diagnosis.1 In the EU, age-adjusted ovarian cancer mortality rates decreased 10% between 2002–12, from 5.8 to 5.2 per 100,000. During this same time period in the USA, the decline was 16%, to 4.9 per 100,000 in 2012.1 However, over the same time period, the prevalence of the disease has increased, with a sharp increase in more recent years (Figure 1).2–4 This increase in prevalence may be attributable to advances in ovarian cancer treatment, which leads to more lines of treatment being given to prolong survival without increasing the rate of cure. The majority of patients with advanced ovarian cancer eventually relapse, so there is a substantial need for new treatments. One potential strategy to reduce the likelihood of recurrence is to use maintenance therapy after chemotherapy to extend the response to treatment and delay the next line of chemotherapy. Such extended treatment is difficult to achieve with chemotherapy due to cumulative toxicities, so other maintenance therapies are needed.
Poly(ADP-ribose) polymerase (PARP) inhibitors are an intriguing new class of therapeutic agents for ovarian cancer. Inhibition of PARP enzymes slows or abolishes the repair of single-strand breaks in DNA and leads to the formation of double-strand breaks.5 Double-strand breaks in DNA are normally rectified through the homologous recombination repair (HRR) pathway.6–8 In cells with homologous recombination deficiency (HRD) such as those with a mutation in BRCA1 or BRCA2 (BRCA), double-strand breaks cannot be efficiently repaired, resulting in cell death via a process termed ‘synthetic sickness’ or ‘synergistic lethality’.9 A therapeutic response to PARP inhibitors has been shown in patients with ovarian tumours with mutations in BRCA or other HRR genes (e.g., RAD51, BARD1, PALB2 and others), and in ovarian tumours with high loss of heterozygosity (LOH), a genomic signature associated with HRD.10–21 However, clinical evidence has emerged showing that patients with ovarian cancer can also receive clinical benefit from PARP inhibitors regardless of their assay-determined HRD status.10,11
Three PARP inhibitors, olaparib (Lynparza®, AstraZeneca, Cambridge, UK), niraparib (Zejula®, Tesaro, a GlaxoSmithKline company, Waltham, Massachusetts, USA) and rucaparib (Rubraca®, Clovis Oncology, Boulder, Colorado, USA) have shown promising results when used as maintenance treatment of recurrent platinum-sensitive ovarian cancer after completion of platinum-based chemotherapy.9–11,13,22–24 In the USA, each of these agents has US Food and Drug Administration (FDA) approval in this setting.25–27 Similarly, in Europe, olaparib, niraparib and more recently, rucaparib are now approved for the maintenance treatment of adult patients with platinum-sensitive relapsed high grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in response (complete or partial response) to platinum-based chemotherapy.28,29 Olaparib is also approved in the USA for the treatment of recurrent ovarian cancer occurring in germline BRCA (gBRCA) mutation carriers who have had three or more prior lines of therapy.26 Rucaparib is approved in both the USA and Europe for the treatment of recurrent ovarian cancer following two prior lines of therapy in patients who have a germline BRCA mutation or in patients who have a somatic BRCA (sBRCA) mutation.27,29
This review summarises the findings from the pivotal clinical trials of olaparib, niraparib and rucaparib that support their use as maintenance therapy for recurrent ovarian cancer. We highlight the key differences in the clinical trial designs and examine the distinct efficacy and safety profiles of each PARP inhibitor.
Key clinical trial data supporting poly(ADP-ribose) polymerase inhibitors as maintenance treatments for recurrent ovarian cancer
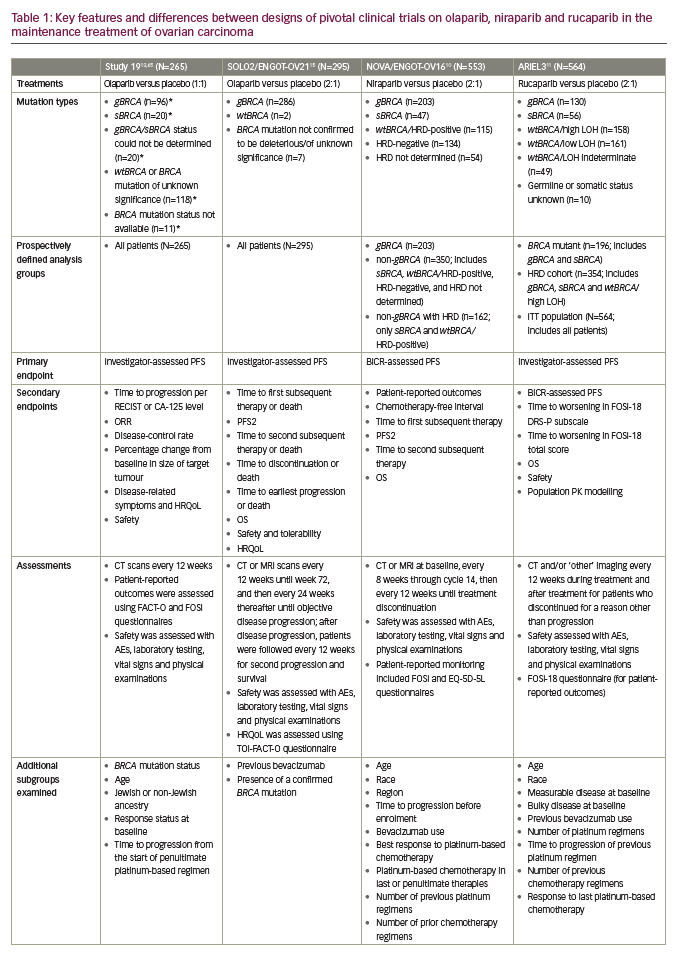
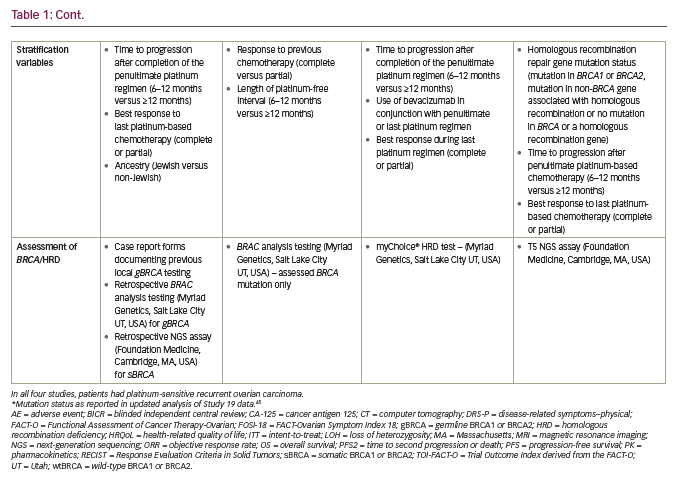
PARP inhibition as maintenance therapy for recurrent ovarian cancer has been investigated with olaparib in Study 19 and SOLO2/ENGOT-OV21,13,15 niraparib in NOVA/ENGOT-OV1610 and rucaparib in ARIEL311 (see end of text for study name definitions). Results from these trials are not directly comparable because of the different study designs and patient populations studied. For example, in NOVA, the primary endpoint was blinded independent central review (BICR)-assessed progression-free survival (PFS), whilst in Study 19 and ARIEL3 the primary endpoint was investigator-assessed PFS. Furthermore, Study 19 and ARIEL3 both examined the primary endpoint in prospectively defined populations that included all patients, whereas NOVA prospectively analysed PFS in distinct subgroups of patients based on the presence/absence of a gBRCA mutation and SOLO2 was limited to women carrying a germline or somatic mutation in BRCA1/2. Notably, in NOVA, patients with a sBRCA mutation were included in the non-gBRCA cohorts, which is unique to this study.10
Differences in the patient populations for each study include the proportion of patients with a germline or somatic BRCA mutation (Study 19, 51%; SOLO2, 97%; NOVA, 45%; ARIEL3, 35%).10,11,13,15 While the phase III NOVA and ARIEL3 trials of niraparib and rucaparib, respectively, included all-comers, the only all-comer data for olaparib in the maintenance setting comes from the randomised phase II trial, Study 19. The proportion of patients with a complete response to prior platinum-based chemotherapy also differed across studies (Study 19, 45%; SOLO2, 46%; NOVA, 51%; ARIEL3, 34%).10,11,13,15 In contrast to NOVA, enrolment in SOLO2 and ARIEL3 was not limited by target lesion size for patients with a partial response to previous platinum-based chemotherapy and thus these trials recruited more patients with bulky residual disease (>2 cm) at baseline (SOLO2, 15%; ARIEL3, 19%).10,11,13,15 An overview of study designs and endpoints for these trials is given in Table 1, key efficacy data are provided in Figures 2 and 3, and a summary of safety data is provided Figure 4. It should be noted that the presentation of data from different studies in these figures is for interest only; the studies included populations with different mutation profiles and had differing inclusion/exclusion criteria and so the results are not directly comparable.
Olaparib
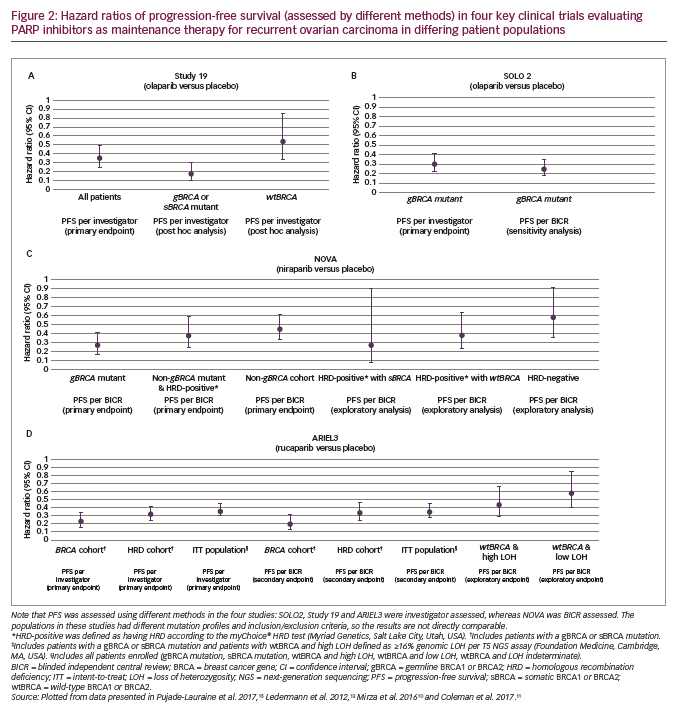
Study 19 was a randomised phase II evaluation of olaparib capsules (400 mg twice daily [BID]) used as a maintenance treatment of patients (N=265) with platinum-sensitive recurrent ovarian cancer.13 The primary endpoint was investigator-assessed PFS from randomisation, which was significantly longer with olaparib than placebo (median 8.4 months versus 4.8 months; hazard ratio [HR], 0.35; 95% confidence interval [CI], 0.25–0.49; p<0.001; Figure 2A). This advantage was seen across all subgroups that were evaluated (BRCA mutation status, age, race [White/non-Jewish], baseline response, and time to progression on penultimate platinum-based regimen). A retrospective analysis of investigator-assessed PFS according to BRCA status, completed after publication of the trial, showed that median PFS in the subgroup with BRCA mutations (gBRCA and sBRCA; approximately 50% of the women) for olaparib versus placebo was 11.2 months versus 4.3 months (HR, 0.18; 95% CI, 0.10–0.31; Figure 2A). In the subgroup with wild-type BRCA (wtBRCA), median PFS was 7.4 months versus 5.5 months (HR, 0.54; 95% CI, 0.34–0.85; Figure 2A).14 For the secondary endpoint of time to progression according to Response Evaluation Criteria In Solid Tumours (RECIST) version 1.130 or cancer antigen 125 (CA-125) level (whichever occurred first), the median was 8.3 months in the olaparib arm compared to 3.7 months in the placebo arm (HR, 0.35; 95% CI, 0.25–0.47).13 The objective response rate (ORR) at 24 weeks (a secondary endpoint) was higher with olaparib (12%; 7/57 patients with measurable disease at baseline) than with placebo (4%; 2/48)13 as was the disease-control rate (secondary endpoint; 53% [72/136] versus 25% [32/129]).31
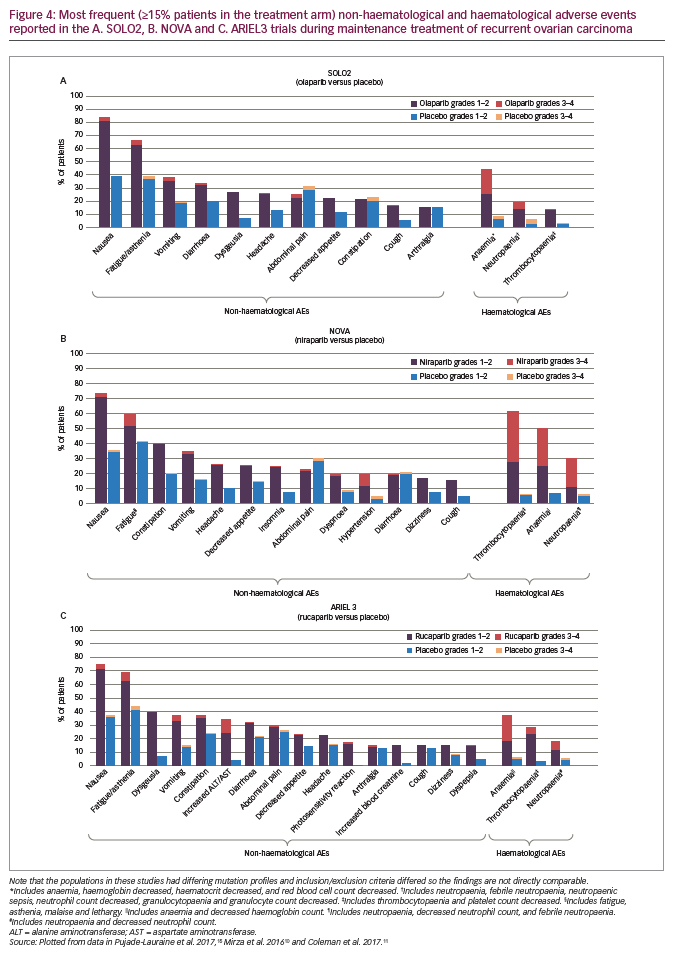
In the overall study population, the most notable adverse events (AEs) were gastrointestinal in nature (nausea [olaparib, 68% versus placebo, 35%] and vomiting [32% versus 14%]) but fatigue (49% versus 38%) was also common. Most AEs were low grade but the commonest AEs of grade ≥3 (olaparib versus placebo) included fatigue (7% versus 3%), anaemia (5% versus 1%), back pain (2% versus 0%), diarrhoea (2% versus 2%), nausea (2% versus 0%) and vomiting (2% versus 1%).13
In Study 19, health-related quality of life (QoL) was assessed using the Functional Assessment of Cancer Therapy-Ovarian (FACT-O) questionnaire total score as well as the Trial Outcome Index (TOI; a subset of 26 FACT-O items) and the FACT-O Symptoms Index (FOSI; a subset of eight FACT-O items). No significant differences were observed between arms in the rates of score improvement across the TOI (odds ratio [OR], 1.14; 95% CI, 0.58–2.24; p=0.7), FOSI (OR, 1.22; 95% CI, 0.60–2.51; p=0.59) and FACT-O (OR, 1.17; 95% CI, 0.60–2.27; p=0.65).32
A more recent long-term follow-up of overall survival (OS) findings in Study 19 (at 79% data maturity) showed a favourable result for olaparib over placebo for OS (HR, 0.73; 95% CI, 0.55–0.95; nominal p=0.021), irrespective of BRCA1/2 mutation status.33 However, this study was not designed to show a statistically significant difference in OS; the p values did not meet the pre-set criterion for significance (p<0.0095) and therefore, the favourable treatment effect reported for OS should only be regarded as descriptive. It is noteworthy that this follow-up showed that 24% of patients received olaparib maintenance therapy for >2 years and 11% received this treatment for >6 years, and this includes patients with and without BRCA mutations. This analysis identified no new tolerability signals during long-term treatment and AEs were generally low grade. The incidence of discontinuations due to AEs was low (6%).
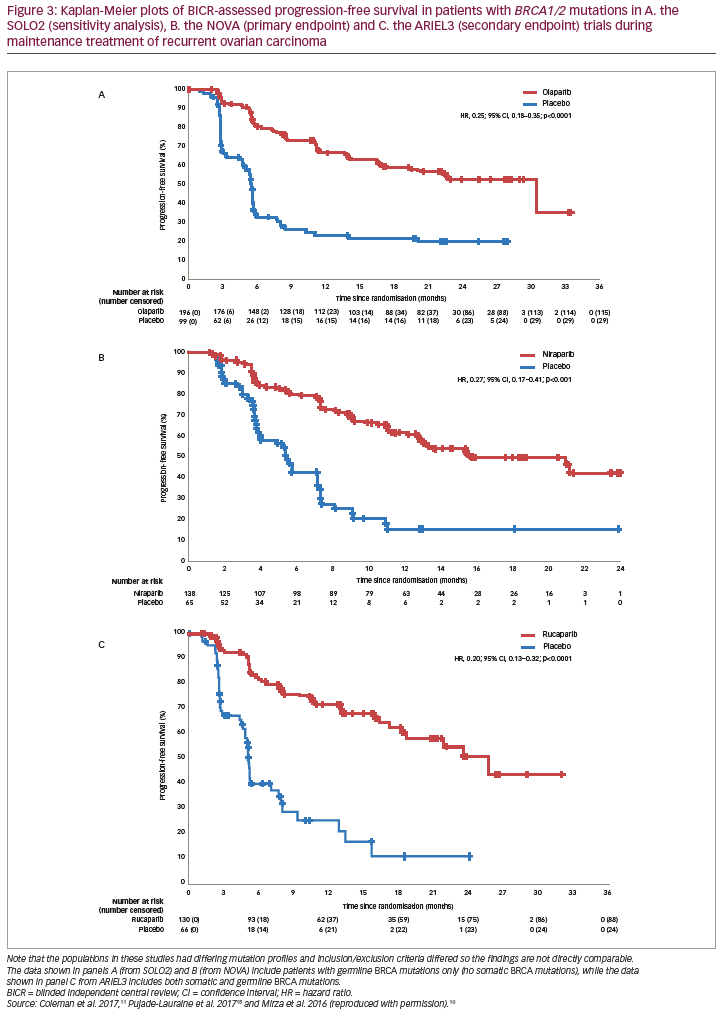
Further pivotal data supporting olaparib in the maintenance treatment of recurrent ovarian cancer comes from the phase III SOLO2 study, in which patients (N=295) with platinum-sensitive recurrent ovarian cancer and a BRCA mutation were treated with olaparib (n=196, 300 mg tablets BID) or placebo (n=99).15 Median investigator-assessed PFS (primary endpoint) was significantly longer with olaparib (19.1 months [95% CI, 16.3–25.7]) than with placebo (5.5 months [95% CI, 5.2–5.8]; HR, 0.30; 95% CI, 0.22–0.41; p<0.0001; Figure 2B). In a sensitivity analysis of PFS by BICR, median PFS was also longer with olaparib (30.2 months [95% CI, 19.8–not reached]) than with placebo (5.5 months [95% CI, 4.8–5.6]; HR, 0.25; 95% CI, 0.18–0.35; p<0.0001; Figures 2B and 3A). Compared to placebo, olaparib improved outcomes for secondary endpoints, including median time to first subsequent therapy or death (TFST; 27.9 months versus 7.1 months; HR, 0.28; 95% CI, 0.21–0.38), median time to second progression or death (PFS2 [a parameter that indicates duration of survival on subsequent therapy following progression on maintenance therapy]; not reached versus 18.4 months; HR, 0.50; 95% CI, 0.34–0.72), median time to second subsequent therapy or death (TSST; not reached versus 18.2 months; HR, 0.37; 95% CI, 0.26–0.53), median time to study discontinuation or death (19.4 months versus 5.6 months; HR, 0.31; 95% CI, 0.23–0.42) and median time to earliest progression or death (16.9 months versus 4.9 months; HR, 0.30; 95% CI, 0.23–0.41).15 Data for the secondary endpoint of OS were immature (24% maturity) with medians not reached for either group (HR, 0.80; 95% CI, 0.50–1.31).15,34
A recent analysis of tumour responses in the SOLO2 study showed an ORR advantage for patients with measurable disease at baseline treated with olaparib versus placebo.35 This advantage was apparent for ORR assessed by investigators (OR, 3.52; 95% CI, 1.34–10.59) and when assessed by BICR (OR, non-evaluable). This same analysis also showed a PFS benefit for patients treated with olaparib versus placebo with either a complete or partial response to platinum-based chemotherapy at baseline. In addition, the analysis determined PFS2 values which revealed long-term benefits of olaparib treatment. This benefit was apparent both for patients with a complete response (HR, 0.41; 95% CI, 0.22–0.77) and for patients with a partial response (HR, 0.57; 95% CI, 0.36–0.91) to platinum-based chemotherapy at study entry.
Most AEs were of grade 1–2 severity, and the most common AEs of any grade (olaparib versus placebo) were nausea (76% versus 33%), fatigue/asthenia (66% versus 39%), anaemia (44% versus 8%), vomiting (37% versus 19%) and diarrhoea (33% versus 20%) (Figure 4A).15 More frequent AEs of grade ≥3 included anaemia (19% versus 2%), neutropaenia (5% versus 4%) and fatigue/asthenia (4% versus 2%) (Figure 4A). Discontinuations due to AEs occurred in 11% of olaparib-treated and 2% of placebo-treated patients. In SOLO2, four patients (2%) in the olaparib arm and four patients (4%) in the placebo arm were reported to have myelodysplastic syndrome or acute myeloid leukaemia. The study showed that the benefits of olaparib on PFS had no detrimental effect on QoL and the toxicities were mostly low grade and manageable. This was emphasised by further analysis of SOLO2 data showing significant improvement in mean quality-adjusted PFS (QA-PFS) for olaparib versus placebo: 14.0 versus 7.3 months (difference, 6.7; 95% CI, 5.0–8.5; p<0.0001).36 In addition, there was also a significant improvement in mean duration of time without symptoms of disease or toxicity (TWiST): 15.0 versus 7.7 months (difference, 7.3; 95% CI, 4.7–9.0; p<0.0001).36
Niraparib
In the phase III NOVA trial (N=553), patients were grouped according to whether they had a gBRCA mutation or a non-gBRCA mutation (this group also included patients with sBRCA mutations) (Table 1). The study also included an analysis that grouped patients by HRD.10 Here, the HRD-positive group included patients with sBRCA mutations or other HRD as determined by the myChoice® HRD test (Myriad Genetics, Salt Lake City, Utah, USA). In the gBRCA cohort, BICR-assessed PFS (primary endpoint) for niraparib versus placebo (n=138 and n=65, respectively) was 21.0 versus 5.5 months (HR, 0.27; 95% CI, 0.17–0.41; p<0.001; Figure 2C and Figure 3B). In the non-gBRCA/HRD-positive subgroup, PFS for niraparib and placebo, respectively, was 12.9 versus 3.8 months (HR, 0.38; 95% CI, 0.24–0.59; p<0.001; Figure 2C). In the overall non-gBRCA cohort, median PFS was 9.3 versus 3.9 months (HR, 0.45; 95% CI, 0.34–0.61; p<0.001; Figure 2C). An analysis of BICR-assessed PFS in subgroups based on HRD status indicated that niraparib treatment was superior to placebo across all subgroups (Figure 2C): non-gBRCA/HRD-positive/sBRCA (n=35 and n=12, respectively): 20.9 versus 11.0 months (HR, 0.27; 95% CI, 0.08–0.90; p=0.02); non-gBRCA/HRD-positive/wtBRCA (n=71 and n=44): 9.3 versus 3.7 months (HR, 0.38; 95% CI, 0.23–0.63; p<0.001); non-gBRCA/HRD-negative (n=92 and n=42): 6.9 versus 3.8 months (HR, 0.58; 95% CI, 0.36–0.92; p=0.02).10,37 Data for secondary endpoints of chemotherapy-free interval (CFI), TFST, PFS2 and OS were subsequently reported.38,39 In the gBRCA cohort, niraparib significantly improved median CFI (22.8 months) compared to placebo (9.4 months; HR, 0.26; 95% CI, 0.17–0.41). Median CFI was also improved in the non-gBRCA cohort (12.7 months versus 8.6 months; HR, 0.50; 95% CI, 0.37–0.67). Median TFST was significantly improved versus placebo for the gBRCA (21.0 months versus 8.4 months; HR, 0.31; 95% CI, 0.21–0.48) and non-gBRCA (11.8 months versus 7.2 months; HR, 0.55; 95% CI, 0.41–0.72) cohorts. Though data were immature (gBRCA, 30%; non-gBRCA, 50%), PFS2 was longer with niraparib than placebo (gBRCA: HR, 0.48; 95% CI, 0.24–0.69; non-gBRCA: HR, 0.69; 95% CI, 0.49–0.96). Less than 20% of OS events had occurred in the overall patient population, but analysis showed a non-significant improvement with niraparib versus placebo (HR, 0.73; 95% CI, 0.48–1.13).38,39
In the NOVA trial, the most frequent AEs of any grade with niraparib versus placebo were nausea (74% versus 35%), thrombocytopaenia (61% versus 6%), fatigue/asthenia (59% versus 41%), anaemia (50% versus 7%), constipation (40% versus 20%), vomiting (34% versus 16%) and neutropaenia (30% versus 6%) (Figure 4B). The most common grade ≥3 AEs with niraparib versus placebo were thrombocytopaenia (34% versus 1%), anaemia (25% versus 0%), neutropaenia (20% versus 2%), fatigue/asthenia (8% versus 1%) and hypertension (8% versus 2%) (Figure 4B). These were managed by modifying or delaying the niraparib dose. With niraparib, 15% of patients discontinued treatment due to an AE compared with 2% with placebo. Myelodysplastic syndrome or acute myeloid leukaemia were reported by five patients (1%) in the niraparib arm and two (1%) in the placebo arm.10
In an analysis of QoL for patients in NOVA, mean pre-progression EQ-5D-5L scores were similar between the niraparib and placebo arms in both the gBRCA (0.838 versus 0.834) and non-gBRCA (0.833 versus 0.815) cohorts.40 At baseline, common symptoms related to QoL included fatigue, pain, nausea, vomiting, bloating and cramps as assessed by the FOSI questionnaire. These symptoms generally remained stable or improved for patients in the niraparib arm, with the exception of nausea which showed an increase at cycle 2 but declined towards baseline levels thereafter.40 A more recent analysis of NOVA study data found that mean TWiST for patients receiving niraparib was 2.95 years for patients with gBRCA mutations compared with 1.34 years for patients without gBRCA mutations.41,42 Niraparib treatment of these patient groups produced PFS benefits of 3.23 years and 1.44 years, respectively, and mean toxicity times of 0.28 years and 0.11 years, respectively. QoL remained stable through niraparib treatment and the pre-progression period compared with placebo.
Rucaparib
ARIEL3 was a randomised phase III trial (intent-to-treat [ITT] population, N=564) to assess the efficacy and safety of rucaparib as maintenance treatment (600 mg BID, n=375) versus placebo (n=189).11 Patients with high-grade, platinum-sensitive ovarian carcinoma were required to have shown an objective response to second-line or later platinum-based chemotherapy. A novel aspect of this trial was the prospective validation of the next-generation sequencing assay performed in collaboration with Foundation Medicine (Cambridge, Massachusetts, USA) to identify tumours with high genomic LOH. In ARIEL3, a cut-off of ≥16% for high LOH was prospectively selected based on the results of a planned post hoc analysis of data from a prior phase II study, ARIEL2.16 In the analysis of ARIEL2 data, a cut-off of ≥16% improved median PFS in the LOH-high subgroup compared to the prespecified ≥14% cut-off (7.2 months versus 5.7 months). The HR for PFS was also improved with the ≥16% cut-off (0.51 [95% CI, 0.34–0.74] versus 0.62 [95% CI, 0.42–0.90]).16
The design of the ARIEL3 study involved a prospectively defined step-down statistical procedure of three nested cohorts.11 Firstly, the BRCA-mutant cohort (n=196) consisted of 130 patients with wt
mutations (n=82 and n=48 for rucaparib and placebo, respectively), 56 patients with sBRCA mutations (n=40 and n=16) and 10 patients with unknown gBRCA or sBRCA status (n=8 and n=2). Secondly, the HRD cohort (n=354) included the BRCA-mutant cohort and a further 158 patients with wtBRCA and high LOH (n=106 and n=52). Thirdly, the ITT population (n=564) consisted of the HRD cohort with an additional 161 patients with wtBRCA and low LOH (n=107 and n=54) and 49 patients with wtBRCA and indeterminate LOH (n=32 and n=17).
The primary endpoint of ARIEL3 (investigator-assessed PFS) showed a significant benefit for rucaparib in each of the three cohorts. The median PFS in patients with BRCA-mutant carcinoma was 16.6 months for rucaparib versus 5.4 months for placebo (HR, 0.23; 95% CI, 0.16–0.34; p<0.0001; Figure 2D). Median investigator-assessed PFS also showed significant advantages for rucaparib over placebo in patients with HRD carcinoma: 13.6 months for rucaparib versus 5.4 months for placebo (HR, 0.32; 95% CI, 0.24–0.42; p<0.0001). In the ITT population, investigator-assessed PFS was 10.8 months for rucaparib and 5.4 months for placebo (HR, 0.36; 95% CI, 0.30–0.45; p<0.0001). Median BICR-assessed PFS for patients with BRCA-mutant carcinoma was 26.8 months for rucaparib versus 5.4 months for placebo (HR, 0.20; 95% CI, 0.13–0.32; p<0.0001; Figure 3C); for patients with HRD, it was 22.9 months versus 5.5 months (HR, 0.34; 95% CI, 0.24–0.47; p<0.0001) and for all patients in the ITT population, it was 13.7 months versus 5.4 months (HR, 0.35; 95% CI, 0.28–0.45; p<0.0001; Figure 2D).11 Pre-planned subgroup analyses found that rucaparib provided a PFS benefit in all clinical subgroups compared with placebo regardless of time to progression on penultimate platinum-based treatment, response to last platinum-based treatment, having a bulky lesion (>2 cm) at baseline and having measurable disease at baseline.11 Investigator-assessed PFS was also significantly longer with rucaparib compared with placebo in patients with wtBRCA/LOH-high carcinomas (median 9.7 months versus 5.4 months, respectively; HR, 0.44; 95% CI, 0.29–0.66; p<0.0001) and in patients with wtBRCA/LOH-low carcinomas (median 6.7 months versus 5.4 months, respectively; HR, 0.58; 95% CI, 0.40–0.85; p=0.0049; Figure 2D). OS data from the ARIEL3 study are currently immature.11
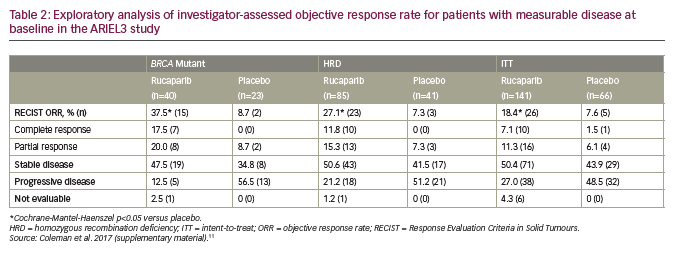
An exploratory analysis of investigator-assessed ORR in patients with measurable disease showed superiority for rucaparib over placebo for all three cohorts in ARIEL 3 (Table 2). These included significant (p<0.05) improvements over placebo for measures of ORR evaluated by RECIST. Among rucaparib-treated patients, there were also substantial improvements over placebo in terms of conversion from a partial to a complete response.11
In ARIEL3, the most common treatment-emergent AEs of any grade (rucaparib versus placebo) were nausea (75% versus 37%), fatigue/asthenia (69% versus 44%), dysgeusia (39% versus 7%), anaemia (37% versus 6%), constipation (37% versus 24%), vomiting (37% versus 15%), increased alanine or aspartate aminotransferase concentration (ALT/AST) (34% versus 4%) and diarrhoea (32% versus 22%) (Figure 4C). Treatment-emergent AEs of grade ≥3 were reported in 56% of patients in the rucaparib group versus 15% in the placebo group. The most notable and frequent of these were anaemia (19% versus 1%), increased ALT/AST (10% versus 0%), fatigue/asthenia (7% versus 3%), neutropaenia (7% versus 2%), thrombocytopaenia (5% versus 0%) and nausea (4% versus 1%) (Figure 4C).11 Discontinuations due to an AE (excluding disease progression) occurred in 13% and 2% of patients, respectively. Myelodysplastic syndrome or acute myeloid leukaemia were reported in three patients (1%) receiving rucaparib, of these, two had gBRCA-mutant carcinoma and one had wtBRCA/LOH-low carcinoma, and no patients in the placebo arm. Overall, the safety findings showed that rucaparib was well tolerated and AEs were manageable; they were mainly low grade and the incidence of more serious events declined after initial cycles of treatment.
In an analysis of the secondary endpoint of time to worsening on the disease-related symptoms–physical subscale of the FOSI-18 questionnaire, no significant difference was noted between the rucaparib and placebo arms in the BRCA–mutant cohort (HR, 1.24; 95% CI, 0.82–1.86).11 An analysis of patient-centred outcomes in the ARIEL3 study has recently reported that mean QA-PFS was significantly longer for patients treated with rucaparib compared with placebo (12.02 versus 5.74 months) and that in patients with wtBRCA, mean QA-PFS was longer for rucaparib than placebo regardless of LOH status.43 Mean quality-adjusted TWiST analysed using all grade ≥3 treatment-emergent AEs was significantly longer for rucaparib than placebo (ITT population, 13.32 versus 6.44 months) and for patients with a BRCA mutation (16.42 versus 6.68 months, respectively). Additional patient-reported outcomes data are expected to be published in the future.
Discussion
For women with platinum-sensitive ovarian cancer, PARP inhibitor maintenance treatment constitutes a new standard of care that can delay the need for further chemotherapy. In key clinical trials, olaparib, niraparib and rucaparib maintenance treatment notably increased PFS for patients with recurrent ovarian cancer following a response to second- or later-line platinum-based chemotherapy.10,11,13,15 As might be expected, in these trials, PARP inhibitors were effective in patients with recurrent ovarian cancer and BRCA mutations. In non-gBRCA patients enrolled in NOVA and in patients with wtBRCA in ARIEL3, niraparib and rucaparib, respectively, were also effective in patients with HRD.10,11 However, while both of these studies found that PFS was improved in patients with tumours associated with HRD, there are distinct differences in the methods used to classify patients with HRD. In NOVA, the HRD status of tumours was evaluated using the myChoice HRD test and the HRD-positive group in the primary analysis included patients with an sBRCA mutation.10 An exploratory analysis in NOVA did show a benefit for patients with wtBRCA and HRD (sBRCA not included). ARIEL3 examined LOH as a biomarker for HRD, and in an exploratory analysis of patients with wtBRCA and high LOH, PFS was improved with rucaparib versus placebo.11 Furthermore, in both studies, patients without a BRCA mutation or HRD demonstrated improvement in PFS, indicating that neither BRCA status nor HRD is a sufficiently precise biomarker to predict which patients will benefit from PARP inhibitor maintenance treatment.10,11 The FDA and European approvals for olaparib, niraparib and rucaparib use as second-line maintenance therapy of recurrent ovarian cancer do not specify BRCA mutation type or HRD status.25–27
The studies described here also demonstrated the benefits of PARP inhibitor maintenance treatment in addition to extension of PFS. In ARIEL3, rucaparib treatment led to a higher ORR than placebo with a number of patients with a partial response converting to a complete response on study.11 In SOLO2, there were also notable advantages in ORR for olaparib compared with placebo in patients with measurable disease at baseline.35 Thus in some patients not only is progression delayed, or even prevented, they may have a deepening of response following maintenance therapy with a PARP inhibitor. Study 19, SOLO2, NOVA and ARIEL3 assessed secondary and exploratory endpoints, including CFI, TFST, PFS2 and TSST (first used in Study 19 as exploratory endpoints).15,38,39,44,45 PARP inhibitor maintenance treatment was associated with improvements in these endpoints, providing further evidence that these drugs are suitable for maintenance therapy in which the objective is to delay the need for additional therapy. Furthermore, the results also suggest that PARP inhibitor treatment does not affect the efficacy of subsequent lines of treatment.15,38,39 For OS, an analysis of all patients in Study 19 showed that olaparib maintenance treatment resulted in a small but non-significant improvement over placebo (29.8 months versus 27.8 months, respectively), with the largest improvement observed in patients with a BRCA mutation (34.9 months versus 30.2 months, respectively).45 A notable minority of patients in this study appear to be long-term survivors with 11% receiving olaparib treatment for >6 years. OS data for the phase III studies are currently immature.10,11,15
The safety and tolerability findings in the pivotal maintenance studies indicate that olaparib, niraparib and rucaparib have somewhat similar AE profiles.10,11,13,15 Many AEs appear to be class effects of PARP inhibitors, such as haematological AEs (e.g., anaemia, thrombocytopaenia, neutropaenia), gastrointestinal AEs (e.g., nausea, vomiting) and fatigue/asthenia. In general, the majority of AEs were low grade and/or transient and were managed with treatment interruption, dose reductions and/or supportive care (e.g., transfusions, antiemetic medications). Discontinuation rates associated with AEs were also similar across trials and were generally higher in the PARP inhibitor arms than in the placebo groups.10,11,13,15
Notable differences in the AE profiles include a higher incidence of any grade and grade ≥3 haematological AEs, particularly thrombocytopaenia, with niraparib compared with olaparib and rucaparib. An exploratory analysis of the NOVA trial (RADAR) identified two significant parameters that could be used to predict the need for niraparib dose modification.46,47 These were a baseline body weight of <77 kg and/or baseline platelet counts of <150,000/µl. It is critical that these criteria be monitored and the dose of niraparib adjusted to improve tolerability. In response to these findings, a protocol amendment was made in the ongoing PRIMA study which permitted starting dose reductions in patients with low body weight or platelet counts.48 This dose modification has recently been reported to be associated with a significant decrease in grade ≥3 haematological and non-haematological toxicities and an approximately 80% reduction in grade 4 thrombocytopaenia and platelet transfusions.49
With olaparib, niraparib and rucaparib it is necessary to monitor full blood counts at baseline and during subsequent treatment due to the reported incidence of myelosuppression and thrombocytopaenia.23,25–27 Hypertension (any grade and grade ≥3) was also observed more frequently with niraparib than with olaparib and rucaparib. Niraparib’s effect on hypertension is believed to be linked to its pharmacological inhibition of the dopamine transporter, norepinephrine transporter and serotonin transporter.50 Niraparib requires monthly monitoring for hypertension during the first year and periodically thereafter during treatment.25,50 With rucaparib, the incidence of any grade elevations in AST/ALT was higher than reported with olaparib or niraparib. These increases were transient and not associated with criteria for drug-induced hepatotoxicity.11 Some of the other differences in the AEs observed may be reflective of the specific PARP enzymes that each drug can target; all three inhibit PARP1 and PARP2 but their actions against other PARPs are variable.23,51,52 Olaparib, niraparib and rucaparib have been reported to increase levels of serum creatinine.25–29,53 This is believed to be a PARP inhibitor class effect resulting from inhibition of creatinine transporters in the kidney but not a result of acute kidney injury.23,54,55 The exact consequences of the variable toxicity of the PARP inhibitors have not been entirely elucidated and may warrant further investigation.
Based on available data from Study 19, SOLO2, NOVA and ARIEL3, maintenance treatment with a PARP inhibitor did not have a detrimental impact on QoL for patients with recurrent ovarian cancer.11,32,36,40 Evaluations of data from SOLO2 and ARIEL3 even demonstrated that olaparib and rucaparib maintenance treatment resulted in a longer period in which patients did not experience disease symptoms or toxicity compared to patients receiving placebo, emphasising another advantage of PARP inhibitor maintenance therapy for patients with recurrent ovarian cancer.36,43
The pivotal studies have demonstrated the utility of PARP inhibition as maintenance treatment following second-line or later platinum-based chemotherapy. In the future, it will be of interest to determine if these, or other PARP inhibitors, could be effectively used earlier in ovarian carcinoma maintenance treatment. The recent SOLO1 phase III study (n=391) investigated the use of olaparib as first-line maintenance treatment in patients with newly diagnosed advanced ovarian carcinoma and germline (n=388) or somatic (n=2) BRCA1/2 mutations.56,57 The results showed clinically significant benefits of PARP inhibitor maintenance treatment versus placebo after a median 41 months of follow-up.57 The risk of disease progression or death was 70% lower with olaparib than with placebo. The AE profile was consistent with the known toxic effects of olaparib. These results indicate the potential of PARP inhibitors as first-line maintenance therapy. A similar phase III randomised study, PRIMA (n=630),48 in which niraparib is being assessed versus placebo as maintenance treatment for stage III or IV ovarian cancer in patients who showed a response to front-line platinum-based chemotherapy, has completed recruitment and the efficacy results are awaited.
Future directions
As use of PARP inhibitors increases and indications expand to allow earlier use, their roles in the treatment of ovarian cancer and sequencing of use relative to other anticancer agents will need to be evaluated. Cross-resistance between PARP inhibitors might be circumvented due to the different PARP enzymes targeted by these therapies, suggesting that a ‘PARP-after-PARP’ strategy may be suitable for patients who have not responded to one of these drugs or has initially responded but then progressed.58,59 This strategy could involve patients who have either stopped PARP treatment after progression or who have stopped after a defined period, for example, following first-line therapy, without progression. One approach that is being investigated in the OReO study (ClinicalTrials.gov identifier: NCT03106987, n=416) is that patients with ovarian carcinoma who originally responded to platinum-based chemotherapy and subsequently progressed on a PARP inhibitor are then switched to olaparib treatment. Patients recruited to this study will have received different previous treatments. Many, for example, will have originally received olaparib and are re-treated with the same drug after further platinum-based chemotherapy. As PARP inhibitors are being introduced earlier in the pathway of treatment, research strategies are needed to explore the effects of re-treatment at a later date. Sequencing in relation to other widely used treatments for ovarian cancer will also require examination as most available data currently comes from trials performed in the second line or later.
The utility of PARP inhibitors in the maintenance setting for recurrent ovarian cancer may be improved by combining them with other agents that have different mechanisms of action, such as those that interfere with DNA replication and repair pathways (e.g., ATR, ATM, CHK1/2 and WEE1).60,61 For example, antiangiogenic agents (e.g., bevacizumab) induce chronic hypoxia in tumours, which induces a down-regulation of BRCA1 and RAD51, leading to HRD, although this effect is controversial.62 Thus tumours with hypoxia-induced HRD may be sensitive to PARP inhibition. This effect was also shown in a recent study using mouse tumour xenografts, in which cediranib treatment resulted in sensitivity to olaparib by producing hypoxia which suppresses the expression of the HRD factors BRCA1/2 and RAD51 recombinase.63,64 However, cediranib also had a direct effect on HRD, independent of its ability to induce tumour hypoxia. This effect was specific to tumour cells and suggested that DNA repair could be manipulated to induce synergistic lethality.
Combination with checkpoint inhibitors (e.g., nivolumab [anti-programmed cell death protein 1 {PD-1}]) is another strategy as tumours with HRD express high levels of novel, tumour-specific protein sequences, which can attract programmed death-ligand 1 (PD-L1)-expressing tumour-infiltrating lymphocytes.65 Preclinical studies have shown that rucaparib combined with an anti-PD-1 inhibitor improved antitumour activity in a BRCA-deficient mouse model.66 Several studies of PARP inhibitors in combination with other agents in patients with ovarian cancer are currently being conducted or are nearing completion. These include PAOLA-1 (ClinicalTrials.gov identifier: NCT02477644, a randomised controlled phase III trial – maintenance olaparib with bevacizumab), OVARIO (NCT03326193, a randomised phase II trial – maintenance niraparib with bevacizumab), ATHENA (NCT03522246, a randomised phase III trial – maintenance rucaparib with nivolumab).67–69 There is also the ongoing VELIA study (NCT02470585, a phase III trial in previously untreated advanced ovarian cancer, randomised to one of three regimens: carboplatin/paclitaxel plus veliparib then veliparib maintenance or carboplatin/paclitaxel plus placebo then placebo maintenance or carboplatin/paclitaxel plus veliparib then placebo maintenance).
An open-label trial involving PARP inhibitors in combination with other treatments for ovarian cancer, the JAVELIN OVARIAN PARP 100 (NCT03642132, avelumab with chemotherapy then maintenance with avelumab and talazoparib), was recently terminated due to futility after another similar study, the JAVELIN OVARIAN 100, failed to meet its primary endpoint.70 A further open-label trial is the ongoing phase III FIRST study (NCT03602859), in which patients are treated with first-line platinum-based chemotherapy with TSR-042 (dostarlimab, anti-PD-1 monoclonal antibody) and niraparib.
An alternative combination approach is being taken in the ongoing ENGOT-OV46/AGO/DUO-O trial (NCT03737643, planned n=927). This is a phase III randomised, placebo-controlled study in which patients with newly diagnosed advanced ovarian cancer will all initially receive durvalumab in combination with chemotherapy and bevacizumab. This will be followed by randomisation to maintenance durvalumab and bevacizumab or maintenance durvalumab, bevacizumab and olaparib; PFS will be the primary endpoint. An additional study of interest is the ENGOT-OV43/BGOG trial (NCT03740165, planned n=1,000) in which patients with ovarian cancer will initially receive a single 3-week cycle of carboplatin/paclitaxel. Subsequently, they will be randomised to pembrolizumab and olaparib, pembrolizumab and placebo or placebo alone, with PFS and OS as the primary endpoints.
Beyond ovarian cancer, the PARP inhibitors have also shown encouraging efficacy in the treatment of a variety of other cancers and are being developed as potential treatments in multiple indications, including haematological malignancies, advanced prostate cancer, pancreatic cancer, bladder cancer, triple-negative breast cancer, squamous cell lung cancer, non-small cell lung cancer, colorectal cancer and metastatic melanoma.71–81 The use of these drugs as maintenance therapy, however, has mostly focused on recurrent ovarian cancer. Future wider use of the PARP inhibitors as maintenance therapy has the exciting potential to delay progression in many other cancer types.
Conclusion
Overall, the use of PARP inhibitors as maintenance therapy is an important development in delaying disease progression in recurrent ovarian cancer, and in Study 19 with the longest follow-up, about 10% of patients have had a sustained response lasting >6 years. The findings from these studies justify further investigation of these agents for use as either monotherapy or in combination with other treatments. The PARP inhibitors have differing properties that could be used to provide increased or more suitable treatment options for numerous patients. Greater awareness of these drugs and wider routine application of them in recurrent ovarian cancer maintenance regimens in the future could improve the prognosis and reduce mortality in this continuingly prevalent and lethal disease in women both with and without BRCA mutations.
Study name definitions
ARIEL3 = Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum-based therapy; NOVA = A trial of niraparib for ovarian cancer that has come back after platinum-based chemotherapy; OReO = Study to examine Olaparib maintenance Retreatment in patients with Epithelial Ovarian cancer; RADAR = Rapid Adjustment of Dose to reduce Adverse Reactions; SOLO1 = Olaparib maintenance monotherapy in patients with BRCA-mutated ovarian cancer following first-line platinum-based chemotherapy; SOLO2 = Olaparib treatment in BRCA-mutated ovarian cancer patients after complete or partial response to platinum-based chemotherapy; Study 19 = a randomised, double-blind placebo-controlled phase II study comparing outcomes with olaparib as maintenance therapy in patients with platinum-sensitive, recurrent high-grade serous ovarian cancer.