Sarcomas are a diverse group of rare muscle, bone and connective tissue tumours. Within soft-tissue sarcoma alone, there are over 70 subtypes, which demonstrate substantial heterogeneity in biology, genetics and clinical behaviour.1 Further complicating their management is the range of treatment responsiveness, with some subtypes wholly refractory to traditional chemotherapy, while others are quite responsive to ifosfamide or anthracycline-based regimens. Yet, even ‘chemosensitive’ subtypes generally demonstrate overall response rates (ORRs) of <50%.2 Targeted therapies have emerged for several diseases, notably imatinib for gastrointestinal stromal tumours (GIST),3 mechanistic target of rapamycin inhibitors for perivascular epithelioid cell tumours,4,5 and anaplastic lymphoma kinase inhibitors for inflammatory myofibroblastic tumours.6 Widespread deployment of targeted agents remains limited by the lack of driver mutations in many subtypes and incomplete biological understanding of others. The range of disease behaviour and treatment refractoriness creates an urgent need for new therapy options.
Immunotherapy has revolutionized the treatment of many malignancies, including melanoma and non-small-cell lung cancer (NSCLC), and holds great promise for sarcoma.7,8 Immunotherapy is predominantly employed through checkpoint blockade using monoclonal antibodies against programmed death 1 (PD1), programmed death-ligand 1 (PD-L1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), in addition to oncolytic viruses and chimeric T-cell therapies. However, immunotherapy has yet to show the dramatic responses in sarcoma achieved in other cancers. The aforementioned heterogeneity leads to varied treatment responses; thus, sarcoma treatment providers are still searching for how to best incorporate immunotherapy into practice. This article outlines the evidence for the effectiveness of various immunotherapy agents as determined by histological subtype and biomarker expression, as well as newer approaches to maximize the deployment of immunotherapy in the care of patients with sarcoma.
Immunotherapy response by histology
The landmark SARCO28 trial (SARC028: A phase II study of the anti-PD1 antibody pembrolizumab [MK-3475] in patients with advanced sarcomas; ClinicalTrials.gov identifier: NCT02301039), a multicentre phase II study of pembrolizumab in advanced soft-tissue and bone sarcomas, was the first to suggest a differential response to checkpoint blockade by histological subtype.9 While 18% of the 40 patients with soft-tissue sarcoma demonstrated objective responses, substantial differences were seen across subtypes: 40% of patients with undifferentiated pleomorphic sarcoma (UPS) had objective responses, compared with 20% in dedifferentiated liposarcoma (ddLPS), 10% in synovial sarcoma and 0% in leiomyosarcoma (LMS). Subsequent expansion cohorts reinforced these patterns, with a 23% ORR among patients with UPS (9/40) but only 10% in the ddLPS cohort (4/39).10
The Alliance A091401 trial (Randomized phase II study of nivolumab with or without ipilimumab in patients with metastatic or unresectable sarcoma; ClinicalTrials.gov identifier: NCT02500797) demonstrated this nuance even more strikingly.11 Patients with advanced sarcoma were assigned to single-agent nivolumab or combination nivolumab and ipilimumab for four cycles, followed by 2 years of nivolumab. Although the results were modest when analysed in aggregate (ORR 5% for nivolumab alone, 16% for nivolumab/ipilimumab), analysis by subtype in the expansion cohorts offered a much different picture. Within UPS, 23% of patients responded to nivolumab alone and 29% to combination nivolumab and ipilimumab, rates that approximate those of first-line therapies.12 In ddLPS, 10.0% of patients responded to nivolumab, and 17.0% responded to combination immunotherapy, outpacing the overall estimate of efficacy. Alternatively, 0 of the 18 patients with GIST and 3 of the 29 patients with LMS (10.3%) responded. These two studies highlight the need for subtype-specific evaluation of immunotherapy response, as response rates among more sensitive histologies, such as UPS and ddLPS, can be otherwise obscured.10,11
Subsequent research has highlighted other immune-sensitive histologies. Alveolar soft-part sarcoma (ASPS) is a rare sarcoma subtype, more common among adolescents and young adults, with consistently high response rates to checkpoint blockade.13 A multicentre phase II study evaluating atezolizumab in 44 patients with ASPS showed an ORR of 37.2%, including one complete response (CR) and 15 partial responses (PR), which were durable for a median of 16.5 months.14 Another 58.1% of patients had stable disease (SD). A multicentre phase II study of geptanolimab (anti-PD1 antibody) had similar findings, with an ORR of 37.8% (14/37), disease control in 86.5% (32/37) and median progression-free survival (mPFS) of 6.9 months.15 Within the French AcSé pembrolizumab study with several cohorts of rare cancers, 50% of patients (7/14) with ASPS responded, with a 12-month PFS rate of 35.7% and 1-year overall survival of 85.7%.16 An evaluation of pembrolizumab combined with vascular endothelial growth factor (VEGF) receptor inhibitor axitinib showed an ORR of 50% (6/12) and a mPFS of 12.4 months.17 While point estimates and specific therapies vary slightly, these findings establish ASPS as one of the most immune-sensitive sarcoma histologies.
Angiosarcoma is a rare, aggressive, endothelial cell malignancy that also appears to be quite sensitive to immunotherapy, although the evidence base is smaller than for ASPS.18 The dual anti-CTLA-4 and anti-PD1 blockade in rare tumours (DART) trial evaluated nivolumab in combination with ipilimumab in a multicentre phase II study across several rare cancers, including angiosarcoma.19 Of 16 evaluable patients with angiosarcoma, four (25%) experienced an objective response. Notably, three of five patients with primary cutaneous disease responded (60% ORR). The 6-month PFS rate was 38% for the entire angiosarcoma cohort. Furthermore, a phase II study of talimogene laherparepvec in combination with pembrolizumab across several sarcoma types showed an ORR of 35%.20 Notably, 3 of 20 patients had angiosarcoma, and two had PRs. Other findings are limited to case series but seem to confirm these results. One series from the University of North Carolina demonstrated objective responses to single-agent pembrolizumab among three of five patients with cutaneous angiosarcoma,21 and another case series from the University of Miami described PRs to a variety of checkpoint inhibitors in five of seven patients with angiosarcoma, most of whom had cutaneous disease.22 Clearly, the early signal is promising for checkpoint blockade with or without talimogene laherparepvec as an effective treatment strategy, particularly among those with cutaneous angiosarcoma.
By contrast, osteosarcoma has consistently been refractory to checkpoint blockade. In the PEMBROSARC study, published in 2019, pembrolizumab was evaluated with metronomic cyclophosphamide in advanced osteosarcomas.23 Of 15 evaluable patients, only four experienced tumour shrinkage, and one had a PR (6.7%); at 6 months, 86.7% of these patients had progressed. Similar findings were seen with nivolumab when evaluated in children and young adults.24 Among 13 patients with osteosarcoma, no objective responses were seen, nor were there any responses among 11 patients with Ewing sarcoma. The apatinib plus anti-PD1 therapy for advanced osteosarcoma (APFAO) trial evaluated apatinib plus camrelizumab (anti-PD1) for advanced osteosarcoma after progression on chemotherapy.25 Objective responses were observed in 9 of 43 patients (20.9%), and 13 were progression free at 6 months (30.2%); thus, these patients did not meet the prespecified endpoints for benefit. Along with LMS and GIST, osteosarcoma represents an immunotherapy-resistant histology.
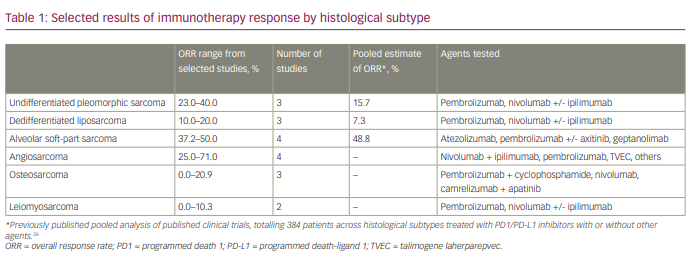
Table 1 shows selected results of immunotherapy responses by histological subtype.26
Predictive biomarkers
Numerous studies have sought reliable biomarkers that predict response to immune activation. In many cancer types, these have offered great promise.27 PD-L1 plays a pivotal role in checkpoint blockade and is widely used as a predictive biomarker. PD-L1 expression is highly correlated with treatment response across the solid-tumour landscape, notably including NSCLC, bladder cancer, gastric cancer and others,28 and the US Food and Drug Administration (FDA) approvals for immunotherapy agents are often contingent upon PD-L1 expression.29 Tumour mutational burden (TMB) reflects the density of mutations within the tumour genome, and high levels of TMB are associated with response to checkpoint blockade across numerous tumour types.30 Similarly, tumours with defects in DNA damage repair pathways (DDR), which are characterized by elevated microsatellite instability (MSI-high), have high TMB and generally demonstrate sensitivity to immunotherapy. This was first shown in colorectal cancer but has since been seen across various tumours.31 Given these findings, the anti-PD1 antibody pembrolizumab received disease-agnostic FDA approval for TMB-high or MSI-high/DDR-mutated tumours.32
Unfortunately, in sarcoma, these biomarkers have not demonstrated the same predictive ability or broad application. In the SARC028 trial, only 3 of 70 evaluable patients had a PD-L1 expression of >1%, although two of those three patients had objective responses.9 Other studies have shown infrequent PD-L1 expression in sarcoma, as low as 0.83% according to a 2018 abstract evaluating 6,100 cases across numerous subtypes.33 Furthermore, PD-L1 expression does not consistently predict response. In patients with ASPS treated with pembrolizumab in combination with axitinib, 52% of evaluable patients expressed PD-L1, but it did not correlate with PFS or response rate.17 Similarly, in the atezolizumab study, 5 of 44 patients had PD-L1 expression compared with 16 responses, without clear correlation.14 In the geptanolimab trial, 30% had a PD-L1 expression of >1%, with no difference in response between those with or without PD-L1 expression.15
The role of other predictive biomarkers in sarcoma is limited by a lack of robust data. In one of the only studies to evaluate TMB in sarcoma, TMB was highly predictive of the response rate to immunotherapy, with ORRs of 16% versus 75% among those with low and high TMB, respectively.34 However, only 5% (4/84) of patients with sarcoma were TMB-high, and these findings have yet to be replicated. Similarly, MSI-high status is uncommon among sarcomas, and its predictive ability has not been evaluated.35 DNA polymerase ε/δ 1 (POLE/POLD1) mutations predict immunotherapy response across cancer types, but are rarely expressed in sarcoma (2.4%), limiting generalizability.36
One promising approach combines several biomarkers into an overarching immune classification.37 Given the important role of the tumour microenvironment in immune response, sarcomas were categorized based on the expression of gene signatures relating to T cells, B cells, tertiary lymphoid structures and biomarkers, including PD1, CTLA-4 and lymphocyte activation gene 3. The authors of the classification identified five distinct sarcoma immune classes from A to E, which predicted responsiveness to checkpoint blockade based on the SARC028 trial population. In class E, ORR was 50%, compared with 25%, 22% and 0% in classes D, C, and A or B, respectively. mPFS was significantly prolonged within class E compared with classes A and B (10 months versus 2 months, p<0.05). The immune class predicted overall survival along the same pattern. These classes track fairly closely with known histological patterns of immune responsiveness, with most LMS falling into class A or B, and most ddLPS defined as class C. Importantly, all histologies were represented in class E, suggesting that this method could discern immune-sensitive tumours better than a histology-only approach. Correspondingly, a range of PD1 expression was observed within class E. The primary barrier to implementation will be replicability. The use of common laboratory methodology in the classification (i.e. immunohistochemical analysis, flow cytometry) allows many institutions to evaluate this approach for broader utility.
Novel approaches
Perhaps the more compelling strategies in leveraging immunotherapy in sarcoma care incorporate additional targeted and cytotoxic agents to improve the efficacy of existing immune treatments, and use specific cancer antigens to target adoptive T-cell therapies. The notion of combination therapy in immuno-oncology has been present since the early trials in NSCLC, which demonstrated robust anti-tumour activity by the combination of checkpoint blockade and traditional cytotoxic chemotherapy, findings that have since been replicated in phase II and III trials.38,39 There is a strong rationale to support the combination of chemotherapy with immunotherapy, as cytotoxic agents may increase antigen presentation by dying tumour cells, promote T-cell activation and inhibit immunosuppressive myeloid cells, among other important immune mechanisms.30–42 Retrospective data in NSCLC have suggested that the combination of chemotherapy and immunotherapy may be more effective than immunotherapy alone, although no randomized trials have evaluated this question to date.43 Numerous on-going sarcoma trials are currently evaluating the efficacy of combination chemoimmunotherapy, including pembrolizumab with eribulin (A phase II study of eribulin and pembrolizumab in soft tissue sarcomas; ClinicalTrials.gov identifier: NCT03899805), trabectedin with dual immune checkpoint blockade (SAINT: A phase 1/2 study of safe amounts of iplimumab, nivolumab and trabectedin as first line treatment of advanced soft tissue sarcoma [STS]; ClinicalTrials.gov identifier: NCT03138161) and an anti-CD40 monoclonal antibody with doxorubicin (A phase II trial evaluating APX005M [a CD40 agonistic monoclonal antibody] in combination with standard-of-care doxorubicin for the treatment of advanced sarcomas; ClinicalTrials.gov identifier: NCT03719430).44–46
There is similar enthusiasm for combining small-molecule inhibitors with immunotherapy. Preclinical data have shown synergistic effects, with inhibition of angiogenesis improving T-cell tumour infiltration and upregulating proinflammatory cytokine expression.47–49 While early trials of the combination of VEGF inhibitors and immunotherapy in renal cell carcinoma (RCC) were limited by toxicity, the combination of axitinib and pembrolizumab has since been shown to be safe and highly effective in advanced RCC.50,51 The combination of VEGF and checkpoint blockade has now become the standard of care for advanced RCC. On-going studies are evaluating this approach in sarcoma, including trials of lenvatinib and pembrolizumab (A pilot study of lenvatinib plus pembrolizumab in patients with advanced sarcoma; ClinicalTrials.gov identifier: NCT04784247), tivozanib and atezolizumab (Atezolizumab plus tivozanib in immunologically cold tumor types; ClinicalTrials.gov identifier: NCT05000294) and cabozantinib with dual immune checkpoint blockade (A randomized phase II trial of cabozantinib combined with PD1 and CTLA-4 inhibition in metastatic soft tissue sarcoma; ClinicalTrials.gov identifier: NCT04551430).52–54
One of the most cutting-edge approaches in sarcoma immunotherapy is the use of adoptive T-cell therapies. By using overexpressed cancer testis antigens (CTAs), researchers have successfully developed genetically modified T cells to specifically target malignant cells. New York-oesophageal squamous cell carcinoma-1 (NY-ESO-1) and melanoma-associated antigen-A4 (MAGE-A4) are outstanding targets for this approach, as they are frequently overexpressed in synovial sarcomas and myxoid/round-cell liposarcomas, among others, while being absent on nearly all normal tissues.55 By harvesting autologous T cells, expanding them ex vivo or modifying the cells to recognize a particular CTA and then returning the cells after the patient has received a conditioning regimen, T-cell therapies have shown promising results. D’Angelo et al. evaluated autologous T cells expressing NY-ESO-1 among 12 patients with synovial sarcoma and found an ORR of 50% – including one CR – a median time to response of 6.2 weeks and a median duration of response of 30.9 weeks.56 Nearly all patients (11/12) experienced adverse events of grade 3 or higher, relating to lymphodepletion (lymphopenia 100%, neutropenia 83%, anaemia 83%). One of the primary risks with adoptive T-cell therapies is the potential for cytokine release syndrome (CRS), in which activated T cells cause a hyperinflammatory state similar to sepsis, risking organ damage and hypoperfusion. Five of 12 patients (41.7%) experienced CRS, although no episodes were fatal and most (3/5) were low grade. Robbins et al. reported similar findings among patients with synovial sarcoma and NY-ESO-1 positivity, with an ORR of 61% (11/18).57 Side effects were similar, with near-universal cytopenias and transient CRS, although frequency and severity were not reported. The 3- and 5-year survival rates were estimated at 38% and 14%, respectively, among patients with an incurable disease who had already progressed through standard therapies.
MAGE-A4 is another CTA in multiple solid tumours that acts to prevent cell-cycle arrest and apoptosis,58 and researchers have seen similar success in leveraging adoptive T-cell therapies in sarcoma. In a phase I dose-escalation study, there were seven PRs (25%) and 11 SDs (39%) among 28 patients; notably, all seven PRs were in patients with synovial sarcoma.59 Adverse effects were again largely due to the conditioning regimen, with high rates of lymphopenia, anaemia and thrombocytopenia. At the 2021 American Society of Clinical Oncology National Meeting, results were presented from the phase II trial (A phase 2 single arm open-label clinical trial of ADP-A2M4 SPEAR™ T cells in subjects with advanced synovial sarcoma or myxoid/round cell liposarcoma [SPEARHEAD-1]; ClinicalTrials.gov identifier: NCT04044768) evaluating the use of MAGE-A4 autologous T cells in patients with synovial sarcoma or myxoid/round-cell liposarcoma.60 Of 25 evaluable patients, there were two CRs (8%), eight PRs (32%) and 11 SDs (44%). At the time of data cutoff, 90% of responders had an on-going response. Over half of the patients (59%) experienced CRS, but 95% of episodes were grade 2 or lower. Cytopenias related to the conditioning regimen were common but largely reversible.
These findings, while early, point to an important role for adoptive T-cell therapies in refractory sarcomas. The overexpression of CTAs on cancer cells but not on normal tissues is a key element of the safety and efficacy of these regimens, providing true targeted anti-tumour immunotherapy. However, there are several limitations to this approach. These modified T cells can only be used among patients who are human leukocyte antigen (HLA)-A*02:01 positive, thus limiting their generalizability.56 Notably, this phenotype is more common among Caucasians, raising equity implications.61 Cell processing, modification and expansion take 1–2 months, a delay that may not be feasible, depending on the clinical circumstances.62 Durability of response remains to be seen – chimeric antigen receptor T-cell therapies in haematological malignancies have shown long-term benefit, but this has yet to be proven in sarcoma.63
Other strategies may potentiate existing therapies and open more treatment options to classically immune ‘cold’ tumours such as sarcoma, including epigenetic modification and downregulation of immune tolerance mechanisms.64 Epigenetic changes allow cancer cells to evade immune recognition, and methylation and histone acetylation have been shown to regulate the expression of NY-ESO-1 antigens.65 Correspondingly, the use of histone deacetylase (HDAC) inhibitors has been shown to increase NY-ESO-1 expression in tumour cells.66 Combining HDAC inhibitors with adoptive T-cell therapies offers a clear potential synergistic benefit, enhancing the expression of cancer antigens, which can then be targeted with effective agents.
Finally, research on resistance to immunotherapy has improved our understanding of immune tolerance, a critical pathway in immune regulation with important implications for therapy. Immune tolerance is necessary for regulating intrinsic inflammatory pathways and preventing harm to normal tissues, yet cancers leverage these same pathways to evade immune detection and activation. Myeloid-derived suppressor cells (MDSCs) are one of several key actors in this process, arising in chronic inflammatory states to protect normal tissues, but are co-opted in malignancy. MDSCs directly inhibit T cells using nitric oxide, reactive oxygen species and cytokines, and seem to promote angiogenesis through the production of VEGF and related peptides, development of metastases and resistance to anti-cancer therapies.67–70 The use of low doses of chemotherapy with gemcitabine and 5-fluorouracil has been shown to eliminate MDSCs, and targeted therapies against the tumour necrosis factor-related apoptosis-inducing ligand receptor may serve as an even more selective approach for inhibition.71–73 Combining MDSC blockade with checkpoint inhibition may be an important mechanism for extending the viability of anti-tumour T cells and enhancing both response rates and durability of benefit.
Conclusions and future directions
While early immunotherapy trials have not led to the rapid sea change seen in melanoma and NSCLC, there is still significant promise in immunotherapy for sarcoma. Histological subtype is a simple way to narrow the breadth of sarcoma pathology to those more likely to respond to immunotherapy, particularly with ASPS, UPS and cutaneous angiosarcoma. Predictive biomarkers may allow further selection of the most sensitive tumours, as well as identify those likely to respond in traditionally immune-refractory histologies. The current standing of PD-L1, TMB and MSI in sarcoma is insufficient to guide this selection. More promising are avenues combining multiple biomarkers to determine sarcoma immune classes and leveraging the CTAs NY-ESO-1 and MAGE-A4 to develop adoptive T-cell therapies.
Ultimately, with a cancer type as varied and challenging as sarcoma, no single strategy will likely be adequate to bring immunotherapy to its full promise. Combination approaches using histology and tumour microenvironment, along with novel strategies targeting epigenetics and immune tolerance will be needed, and predictive biomarkers may ultimately differ between histological subtypes. However, caution must be taken to ensure any new stratification approach is widely applicable for patients across the USA and globally.
Finally, we need more active immunotherapy agents in sarcoma. Given the promise of checkpoint blockade, the current wave of clinical trials is evaluating mechanisms to prime sarcomas to improve immunogenicity, whether in combination with chemotherapy, tyrosine kinase inhibitors or other mechanisms. Improved therapies, more precise predictive biomarkers and continued histological stratification are needed to fully realize the promise of immunotherapy in sarcoma.