Tenosynovial giant cell tumor (TGCT)—previously referred to as giant cell tumor of the tendon sheath or pigmented villonodular synovitis (PVNS)—is a largely benign, rare, proliferative lesion arising from the synovial lining of joints, bursae, and tendon sheaths.1 This review serves to discuss the epidemiology, molecular biology, clinical behavior, and the current treatment modalities of TGCT that are being effectively utilized for patients with a specific focus on CSF1R inhibitors; namely, the newly US Food and Drug Administration (FDA)-approved pexidartinib.
Epidemiology
The incidence of TGCT depends somewhat on subtype: localized TGCT has an incidence of 30.3 per million person-years, while diffuse TGCT is incident at 8.4 per million person-years.1–4 The mean age at diagnosis approaches 47 years; most cases present between the third and sixth decades of life, and the tumor exhibits a slight female predominance.5 In 2013, the World Health Organization (WHO) classified TGCT into clinically distinct localized (TGCT-L) or diffuse types (TGCT-D), although both types are microscopically and genetically identical.6,7 TGCT subtypes are associated with anatomic location: localized tumors (lobulated, well-circumscribed lesions) are more common in the digits of the hand, while diffuse tumors (aggressive lesions involving part or all of the synovial lining) are typically seen in large joints, most commonly the knee, followed by the hip.2,7,8 Although TGCTs are chiefly benign, malignant TGCTs do exist. A malignant TGCT is extremely rare, with fewer than 50 cases reported in the literature, and has a poor prognosis, with median survival of 22.5 months after diagnosis, despite aggressive treatment.4,9,10
Molecular biology
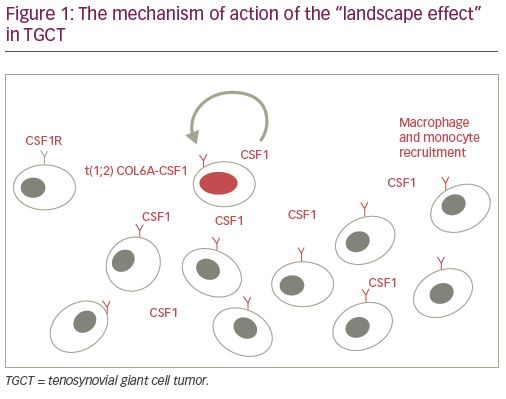
Upon initial descriptions of the lesion, the etiology of TGCT was thought to be related to trauma and inflammation.8,11,12 The notion that TGCT is, in fact, a neoplastic process instead of an inflammatory one was first made in 1984 when it was determined that monoclonal proliferation of fibroblasts and histiocytes contribute to tumor formation.13 Identification of genomic aberrations, including aneuploidy, chromosome-1 deletion, and trisomy 5 and 7, allowed for definitive genetic characterization and confirmed its neoplastic nature.14–19 A translocation involving the short arm of chromosome 1p11-13, with resultant hyperexpression of macrophage colony-stimulating factor 1 (CSF1), has since been shown to contribute in large part to the development of TGCT.20 The CSF1 gene is located at the 1p13 breakpoint, and the translocation leads to a classical fusion event between the collagen 6A3 (COL6A3) promoter element and CSF1. Although only a small percentage (2–16%) of cells in TGCT carry the t(1;2) translocation, excessive resultant CSF1 secretion attracts monocytes and macrophages due to their expression of the CSF1 receptor (CSF1R); this phenomenon is dubbed the “landscape effect” (Figure 1).20,21 Two subtypes of cells are thought to contribute to said “landscape.” One group exhibits both CSF1 overexpression and CSF1 translocation, while the second group lacks the CSF1 fusion event, and is surmised to carry other genetic rearrangements altering CSF1 regulation, ultimately leading to high CSF1 mRNA/protein levels.20,21 The recruited inflammatory cells differentiate and subsequently create the neoplastic multinuclear landscape that is characteristic of TGCT.
Clinical presentation and diagnosis
The clinical presentation of TGCT varies based on whether the tumor is localized or diffuse, and whether it arises intra-articularly or within a tendon sheath. TGCT-L most commonly presents as a painless, slowly growing soft tissue swelling on the hand near the interphalangeal joints of the digits on the palmar side, while TGCT-D occurs most commonly intra-articularly in the knee (75% of cases), followed by the hip.2,8,22 Most TGCT-D lesions are painless (one review of 50 cases of TGCT-D found that only 14% present with pain).23 This lesion may present with discomfort, swelling, stiffness, and locking (“pseudomeniscal” symptomatology if in the knee) of the involved joint.5 Additionally, TGCT-D may present with bloody effusions, frank hemarthroses, arthritis-like symptoms including crepitus, visible swelling, and palpable nodularity of the affected joint. It is not uncommon for a patient with knee involvement to present with recurrent knee effusions in the absence of related trauma over a long period of time.
Symptoms also differ based on whether the tumor is extra- or intra-articular. The extra-articular soft tissue forms commonly involve tendons around the thigh, hand, and foot. The extra-articular and tendon sheath forms present with a very slowly progressive and painless mass, which may cause skin tension in the fingers or toes. These patients may present with gradual onset of uncomfortable or no-longer-fitting footwear. The intra-articular forms of TGCT-D present with discomfort and repeated swelling with restricted range of motion.1
Physical exam of TGCT-L often reveals a soft, palpable mass in superficial locations overlying the involved joint, while TGCT-D often presents with pain and swelling. The mass in both cases is sometimes associated with heat and recurrent periarticular effusion or edematous swelling. Though non-specific, these physical examination findings in the appropriate clinical setting warrant further workup, beginning with imaging.
Imaging
Radiography
Radiography is the appropriate first-line modality for imaging workup of localized joint pain, and over 30% of TCGT-D will present with osseous abnormalities. Bony abnormalities are most likely to occur in the hand (TGCT-L) and hip (TGCT-D), which are not as capacious as the shoulder and knee. Finger erosions and other bony erosions seen in TGCT-L are focal regions due to longstanding pressure on the periosteum causing the bone to recede with well-corticated margins versus the multiple, destructive, infiltrative erosions without cortication seen in TGCT-D. These cystic, erosive bone lesions are thought to occur due to hyperplastic villous formations extending through vascular foramina into cortical bone. In contrast to degenerative subchondral cysts of osteoarthritis, erosions related to intra-articular lesions in TGCT-D tend to occur in the non-weight-bearing regions, at the capsular insertion, and do not calcify (in contrast to synovial chondromatosis).1
Magnetic resonance imaging
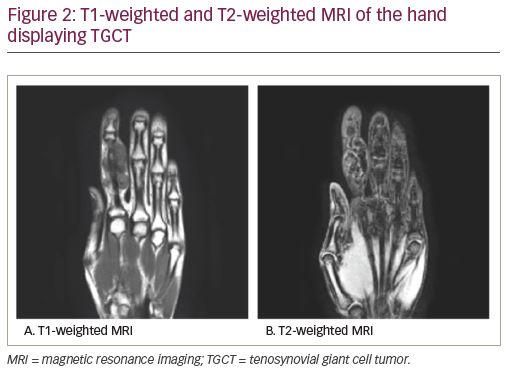
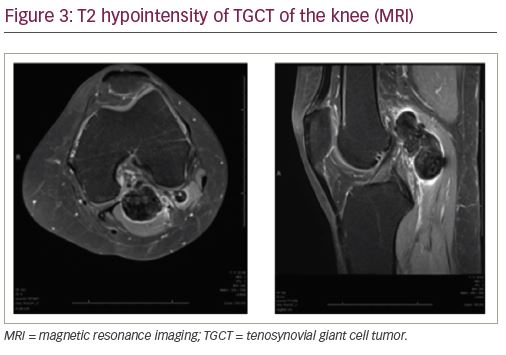
Magnetic resonance imaging (MRI) is highly sensitive for diagnosing TGCT and is considered the definitive imaging modality for diagnosis and surgical planning due to soft tissue detail and resolution.1,7 While T1-weighted images are often non-contributory because the tumor blends in with adjacent synovial fluid (Figure 2a), T2-weighted images often depict a heterogeneously low signal intensity due to the dense collagen and hemosiderin-laden macrophages present within the tumor (Figure 2b, Figure 3). Hemorrhagic effusion, coupled with hemosiderin deposition and concentration in xanthoma cells, can be exploited diagnostically by observing “blooming” artifact on gradient-echo sequences, whereby the paramagnetic hemosiderin molecule causes dephasing of adjacent nuclei and produces a pronounced local signal void.7,24 MRI is used to diagnose TGCT and to help determine the degree of local anatomic involvement to decide if open, arthroscopic, or combined approaches may be utilized to best manage the tumor with respect to the expected morbidity of the surgery.
Other modalities
While ultrasound may be used to assess clinically palpable soft tissue masses, it primarily serves as an image-guidance modality for percutaneous needle biopsy in the management of patients with TGCT. Doppler ultrasound can be utilized to display hypervascularized lesions and can optimally guide synovial biopsy.1,7 While distant staging plays no role in the vast majority of cases, incidental detection of TGCT during staging positron emission tomography-computed tomography (PET-CT) for other cancers has been reported in a small percentage of patients, and TGCT should be among the top considerations when an intra-articular metabolically active mass is identified on PET-CT.25
Pathology
Characteristic histologic players in TGCT include a polymorphous population of mononuclear stromal cells, multinucleated, osteoclast-like giant cells, macrophages, siderophages, hemosiderin depositions, epithelioid histiocytes, foam cells, and/or lymphocyte clusters.7 Though clinically dissimilar, TGCT-D and TGCT-L exhibit identical histologic composition. Behavior is what can set the two apart under a microscope: TGCT-D differs in that it may aggressively entrap and infiltrate adjacent soft tissue and may cause bony erosion. In addition, pseudoalveolar spaces and cellular areas without giant cells may be present.4,23 Lastly, it is possible to see extra-articular foci in the diffuse type, in which case the synovium ultimately appears brown and is diffusely covered by villous and coarse nodular outgrowths.
Standard of care and unmet needs
Standard of care for the treatment of both TGCT-L and TGCT-D is surgical excision of the lesion, either arthroscopically or with open resection. TGCT-L is favorably treated with surgical resection alone, with recurrence rates <6% on average.26 Higher recurrence rates are observed after resection of TGCT-D, at >50% depending on the surgical procedure and follow-up time, representing an unmet need regarding management of this tumor.2 It is critical to assess for resectability because the primary treatment option for TGCT-D is surgery. However, if surgery is not feasible or there is recurrence, systemic therapies are considered in the form of CSF1 inhibitors (nilotinib, imatinib, pexidartinib, emactuzumab, cabiralizumab, and MSC110). It is important to note that there are few data on long-term outcomes due to the novel role of these agents in treating TGCT.27
Management strategies
Role of surgery
Careful and complete excision using an arthroscopic or open approach often achieves surgical cure in the management of TGCT-L. In fact, a review on arthroscopically treated TGCT-L reported no recurrences in patients followed up to 4.5 years postoperatively.26 Traditionally, treatment of TGCT-D of the knee involved combined anterior arthroscopic and open posterior excision and reports the most favorable outcomes.28 Arthroscopic techniques continue to improve, and some recent literature has advocated the use of arthroscopic total synovectomy (denotes synovectomy of the posterior compartments as well as the anterior compartments) given a decrease in morbidity of the procedure compared to an open approach (PMID 29354472).29 In the case of extra-articular foci, combined arthroscopic and open synovectomy are recommended with recurrence rates of 8–18%, and up to 50% in some studies.3,4,30,31 Intra-articular TGCT recalcitrant to various treatments or late presentation often causes symptomatic arthritis of the joint, which can be treated surgically with arthroplasty/replacement of the involved joint.
Role of radiation therapy
To minimize recurrence, postoperative radiation can be utilized as adjuvant therapy, but it is typically only used in refractory cases. Some reports have shown that adjuvant radiation reduces local recurrence of TGCT-D to as low as 4%.32 Injectable intra-articular radionuclides have been described for use as an adjunct to surgical treatment of TGCT-D, but any evidence of benefit is inconclusive.33 It should be noted that adjuvant radiation therapy (both traditional and injectable types) can cause irreversible side effects such as fracture, fibrosis, edema, and joint stiffness.34
Role of medical treatment
As each etiologic target of TGCT is identified, medical treatment of the tumor has begun to take on an increasingly relevant and important role. Given that much of the pathogenesis of the tumor has been attributed to hyperactive CSF1 binding to CSF1R in macrophages and monocytes, monoclonal antibodies have been developed to combat CSF1R activation with the aim of preventing neoplastic regulation at the cellular level (Figure 4). Drugs currently being studied include CSF1R inhibitors cabiralizumab, emactuzumab, imatinib, nilotinib, and of special importance, pexidartinib.35,36 A 2008 study reported a case of complete remission of recurrent TGCT of the right elbow in a 34-year-old woman after treatment with imatinib, and another study demonstrated reduction in the size of recurrent hip TGCT with the same approach.37,38 A 2010 study displayed in a xenograft TGCT model that anti-CSF1 antibodies inhibit macrophage infiltration into the tumor. Multiple clinical trials are investigating the use of CSF1R inhibitors to treat TGCT, and one CSF1R inhibitor passed phase III clinical trials to achieve FDA approval.9,39
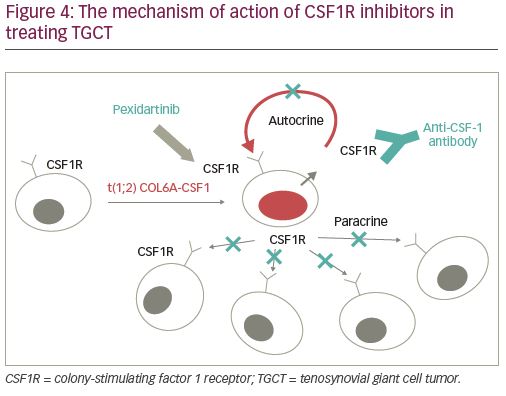
Pexidartinib (Turalio®, Daiichi Sankyo, Chuo, Japan), an oral tyrosine kinase selective inhibitor of CSF1R, is distinct from the rest in that it is the first systemic therapy with FDA approval (in August 2019) in the USA for the treatment of TGCT.40 It targets CSFR1, KIT proto-oncogene receptor tyrosine kinase (KIT), and FMS-like tyrosine kinase 3 (FLT3) harboring an internal tandem duplication (ITD) mutation, overcoming the inherent ability of the tumor to overproduce CSF1.40,41 Pexidartinib is indicated in the USA for use in patients specifically with symptomatic TGCT associated with severe morbidity and significant functional limitations, and is not amenable to improvement with surgical intervention.40,41 The ENLIVEN study, an international phase III study of pexidartinib versus placebo, displayed a robust tumor response with improved patient symptoms and functional outcomes after treatment with pexidartinib at week 25 by RECIST (absolute difference 39%).39 Patient Reported Outcomes Measurement Information System (PROMIS) data report on patients’ ability to perform activities of daily living, and scores were almost identical between the pexidartinib and placebo groups at 37.5 (standard deviation [SD]: 4.9) and 38.9 (SD: 6.1), respectively. Range of motion, reported as percentage of reference range of motion, was also comparable between the two groups, at 62.5 (SD: 24.8) and 62.9 (SD: 21.8), respectively. The overall response rate by tumor volume score—a score that calculates tumor volume as a percentage of the entire synovium and in which a partial response is designated by at least a 50% tumor volume score decrease from baseline—displayed 64% of patients treated with pexidartinib as having at least a partial response to treatment.42 Although impressive, the trial had a relatively small sample size (n=61 in the pexidartinib group and n=59 in the placebo group), and identified serious adverse events in 13% of the pexidartinib group compared to 2% of the control group, reporting significant hepatotoxicity as a drug-related adverse event.39 The study concluded that pexidartinib may be considered to treat TGCT when the tumor is impervious to surgical management.39 There may be an expectation of indefinite continuation of the medication in the setting of recurrent or unresectable disease, which may have unknown complications in the long term based on the limited data we currently have. A future approach to the treatment of patients with TGCT-D may involve a combination of surgery and systemic therapy with CSF1R inhibitors.
There are multiple upcoming clinical trials involving CSF1R inhibitors, including an open label phase I/II study of DCC-3041 (Clinicaltrials.gov Identifer: NCT03069469; Decipheral Pharmaceuticals Inc., Waltham, MA, USA), which is currently recruiting; a phase I/II study of cabiralizumab in patients with TGCT-D, which has completed recruiting (Clinicaltrials.gov Identifier: NCT02471716); and a study of pexidartinib in participants with moderate hepatic impairment compared to healthy participants, which also is currently recruiting (Clinicaltrials.gov Identifier: NCT04223635). The latter trial highlights one of the more difficult aspects of using pexidartinib, as blood draws must be taken weekly for 8 weeks to monitor for hepatic dysfunction.
Conclusion
In summary, TGCT is a rare and benign, yet locally destructive, tumor of the synovia, joints, bursae, and the tendon sheath. TGCT-D is often difficult to treat solely with surgery, thereby warranting the study of additional treatments. Imaging plays a critical role in defining the extent of disease and determining resectability. Recent identification of an oncogenic translocation that upregulates CSFR1 in these tumors provides a rational molecular target for medical therapy, and CSFR1 inhibitors appear to be beneficial in treating unresectable diffuse types. Further studies are warranted to determine whether CSFR1 inhibitors may be used neoadjuvantly or adjuvantly with surgery, and to determine the effectiveness of these systemic therapies in decreasing long-term recurrence risk.