
Phenoxodiol ([2H-1-Benzopyran-7-ol, 3-(4-hydrophyphenyl] (PXD) is a synthetic derivative of genistein, a naturally occurring plant isoflavone. In the 1980s, genistein was discovered to have modest anticancer activity, and alterations in its structure created PXD and increased its anticancer activity without any toxicity. In vitro and in vivo studies showed PXD not only to be directly cytotoxic to a range of cancers, but also to sensitize chemoresistant ovarian cancer cells to platinums and taxanes.1 Recently, PXD was granted ‘fast track’ status by the US Food and Drug Administration (FDA) in its development as a chemosensitizer for platinums and taxanes for the treatment of recurrent ovarian cancer. This article summarizes what is known about PXD’s mechanism of action and the clinical studies employing this compound.
Pre-clinical Studies
Initial studies with PXD published in 2002 described its ability to inhibit the catalytic activity of DNA topoisomerase II.2 One year later, one of the first sets of data supporting its anticancer activity showed that it inhibits dimethylbenz(a)anthracene-induced mammary carcinogenesis in female Sprague–Dawley rats.3 The molecular mapping of PXD’s mechanism of action was initially performed in ovarian cancer cells. Kamsteeg et al. showed that PXD is able to induce apoptosis in chemoresistant ovarian cancer cells both in vitro and in vivo at doses that are not toxic to normal ovarian surface epithelial cells.4 They showed that PXD is able to activate caspases, inhibit the FLICE-inhibitory protein (FLIP) and X-linked inhibitor of apoptosis (XIAP), which are potent apoptotic inhibitors, and, more importantly, chemosensitise ovarian cancer cells to Fas-induced apoptosis.
A more thorough characterization of the PXD-induced apoptotic pathway was later described, also using ovarian cancer cells. Alvero et al. showed that PXD induces an early activation of the apoptosis-initiating molecule caspase-2 with concomitant proteasome-dependent degradation of the apoptosis inhibitor XIAP.1 They also showed that in addition to previous reports showing that PXD sensitizes ovarian cancer cells to Fas-mediated apoptosis, it is able to sensitize chemoresistant cells to a wide range of chemotherapy agents, including paclitaxel, carboplatin, gemcitabine, docetaxel, and topotecan.1,5,6 PXD’s antiproliferative activity was studied in head and neck squamous carcinoma. Aquero et al. showed that PXD is able to induce caspase-independent clonogenic death in these cells. They further showed that PXD is able to induce the expression of cyclin-dependent kinase 2 (CDK2) and therefore induce G1-S arrest, and that this occurs as a result of p53-independent p21 induction.7 In addition to its cytotoxic properties, PXD has also been shown to exhibit antiangiogenic properties. Using human umbilical vein endothelial cells, Gamble et al. showed that PXD inhibited endothelial cell proliferation, migration, and capillary tube formation.8 More importantly, they also showed that PXD is able to inhibit the expression of the matrix metalloproteinase (MMP)-2, which is a major matrix-degrading enzyme and is therefore important for the process of metastasis.
The sphingomyelin pathway was investigated in the search for an upstream mediator of PXD-induced toxicity in cancer cells. Sphingosine-1-phosphate (S1P), a product of sphingosine kinase (SK), has been shown to modulate a number of cell signaling processes, including the Akt pathway, resulting in the suppression of apoptosis.9 Conversely, ceramide, the product of sphingomyelinase, has been well characterized as an inhibitor of cell growth, a promoter of G1 cell cycle arrest, and an inducer of apoptosis.10 PXD inhibited the production of S1P and induced the formation of ceramide in the HeLa cells (immortalized cervical cancer cells).11 Furthermore, at the 2005 ASCO Annual Meeting, Cabot et al. showed that PXD promoted the formation of ceramide in head and neck (KB-3-1), ovarian (A2780), and adriamycin-resistant breast cancer (MCF-7-Adr). The demonstration that PXD is able to upregulate ceramide in the process of cell death induction correlates well with data showing that PXD activates caspase-2,1 since one of the upstream regulators of caspase-2 activation is ceramide.
More recent studies have identified redox molecules as potential targets for PXD action. Fine control of the intracellular redox status of cells has been shown to be critical for cell viability, growth, and function.12 Perturbation of key redox determinants such as the NADH/NAD+ ratio, glutathione balance, and membrane ubiquinone redox status and content can have profound effects on cells resulting in apoptosis. Scarlett et al. suggested that because of its hydrophobic character PXD may have a tendency to partition in membrane systems, and could therefore disrupt essential redox cycling at many different levels within cells and their membranes.12
PXD was recently shown to bind with high affinity to a purified recombinant NADH-oxidase, compromising its ability to oxidize both NADH and ubiquinol and to catalyze protein disulfide–thiol interchange activity.13 This oxidase is a truncated form of a tumor-specific cell surface NADHoxidase (tNOX), which is thought to be involved in the transfer of electrons from intracellular NADH to an extracellular acceptor via plasma membrane ubiquinone.13
Clinical Studies
There have been eight clinical studies in which patients have been treated with oral PXD.
Clinical Study 1
In the phase Ib/IIa safety and efficacy study of PXD in males with hormonerefractory prostate cancer (HRPC) trial, 30 patients enrolled in a treatment cycle of 28 days where PXD was given every eight hours for 21 days at dosages of 20, 80, 200, or 400mg and, prior to a protocol amendment, two patients were enrolled at 360mg per dose. Patients received six treatment cycles (six months) or were treated until there was disease progression. At six months, those patients considered by the principal investigator to be deriving a clinical benefit were continued on PXD therapy.
Twenty-five evaluable patients with HRPC have been studied. Six patients were treated in 20 and 80mg dose strata, five at 200mg dose levels, and eight at the 400mg dose level. There were no drug-associated toxicities or intolerance in any dose strata for the six months of treatment. The eight-hourly dosing regimen resulted in steady-state plasma levels of drug with no accumulation over time. The mean steady-state PXD levels in the 20, 80, 200, and 400mg dose strata were 1.57, 8.14, 16.31, and 27.19μg/ml, respectively. The clinical outcomes of the study are summarized in Table 1.
Clinical Study 2
In a phase IIa evaluation of oral PXD in patients with hematological cancers, seven patients were evaluated with oral PXD every 12 hours over 42 days with an intended dose escalation every 14 days. The lowest dose stratum was 13mg/kg and the highest dose stratum was 58mg/kg. The dosages used and duration of treatment are shown in Table 2. There were no drug-associated toxicities or intolerance in any dose strata with PXD. No evidence of tumor response was observed, and the study was terminated due to the inability of the blood to deconjugate glucuronide or sulfate moieties of PXD. Combined with the high level of susceptibility of PXD to conjugation, this made hematological cancers an inappropriate clinical target for PXD.
Clincal Study 3
In a phase IIa safety and efficacy study of the neoadjuvant use of oral PXD in women with a primary diagnosis of squamous cell carcinoma or adenocarcinoma of the cervix, vagina, or vulva, patients were treated with PXD 50 (n=6), 200 (n=10), or 400mg (n=10) eight-hourly for 28 consecutive days on a neoadjuvant basis prior to surgical resection; 26 patients completed treatment. Tumor response was assessed over the 28-day treatment period by both a change in tumor burden using the Response Evaluation Criteria in Solid Tumors (RECIST) and by histochemical evidence of a biological effect (e.g. apoptosis). No incidences of PR have been recorded (against RECIST criteria), although many patients have shown a reduction in the sum longitudinal diameter of between 20 and 27%. Of the six patients treated with PXD 50mg, the incidence of stabilized disease (SD) and disease progression (DP) was two and and four, respectively. Of the eight patients treated with PXD 200mg, the incidence of SD and DP was eight and zero, respectively. The data for the 400mg stratum have yet to be collected. This trend suggests an antitumor effect of PXD in this setting, a fact supported by histological evidence showing an increase in apoptotic index and decrease in mitotic index in the 200mg stratum. There was one1 serious adverse event (SAE) deemed definitely related to PXD: prolonged bleeding time in a patient on the 400mg arm. The data are currently being cleaned and will be available for analysis shortly.
Clincal Study 4
In a current phase Ib safety and efficacy study of PXD in combination with cisplatin or carboplatin in patients with advanced or metastatic cancer not suitable for surgery or radiotherapy, 25 patients were treated with a oral PXD in combination with cisplatin or carboplatin. The primary objective of this study was to determine the recommended phase II dose of PXD in combination with cisplatin or carboplatin. Patients were given oral PXD 50 (n=6), 100 (n=6), 400 (n=6), or 800mg (n=7) every eight hours daily for 10 consecutive days followed by 11 days of rest. In addition, patients received either cisplatin 50mg/m2 on days two and nine of each 21-day cycle or carboplatin area under the curve (AUC)=5 on day three every 21 days. The dose-limiting toxicity (DLT) was diarrhea at the 800mg dose level and the maximum tolerated dose (MTD) was determined to be 400mg orally every eight hours. In this trial there were four serious adverse events: two cases of diarrhea, one case of ventricular arrhythmia, and one case of vomiting. All of these patients also received cisplatin therapy. The data are being analyzed for this study and the report is being prepared.
Clincal Study 5
In a phase II evaluation of PXD in combination with cisplatin or paclitaxel in women with a history of platinum/taxane-refractory or -resistant epithelial ovarian, fallopian tube, or primary peritoneal cancers, patients with recurrent (n=60) late-stage epithelial cancer are being treated with weekly intravenous (IV) PXD 3mg/kg or oral PXD 400mg every eight hours in combination with IV weekly carboplatin (AUC=2), IV weekly cisplatin (40mg/m2), and paclitaxel (80mg/m2) IV weekly (six arms). Initial results were encouraging, with an objective response rate of 30 and 14% for the PXD plus cisplatin and PXD plus paclitaxel groups, respectively, and an overall disease control rate of 75% for PXD plus cisplatin and 76% for PXD plus paclitaxel. There were eight serious adverse events reported in this trial: skin rash (n=1), diarrhea (n=1), pancytopenia (n=1), peripheral sensory neuropathy (n=1), thrombocytopenia (n=1), hypersensitivity reaction (n=1), neutropenic sepsis (n=1), and febrile neutropenia (n=1). The data are currently being analyzed.
Cinical Study 6
A multicenter, randomized, double-blind phase III efficacy study is currently under way comparing PXD (oral dosage form) in combination with carboplatin versus carboplatin with placebo in patients with platinum-resistant or platinum-refractory late-stage epithelial ovarian, fallopian, or primary peritoneal cancer following at least second-line platinum therapy. This global phase III trial of 340 patients will expand on phase II clinical evidence that PXD has the ability to reverse chemoresistance to platinum drugs in late-stage ovarian, fallopian, and primary peritoneal cancers.
Patients are those who have received at least two lines of platinum therapy (cisplatin or carboplatin) and found that their tumor is platinum-resistant (disease progression within six months of completion of a platinum course of therapy) or platinum-refractory (disease progression while on platinum therapy). This cohort of patients are unlikely to respond to further treatment with a two-, three-, or four-weekly platinum regimen, but may be responsive to a more dose-dense therapy regimen of weekly platinum. The treatment arms are PXD two 200mg capsules every eight hours plus weekly carboplatin AUC=2 for four weeks or weekly carboplatin AUC=2 plus two oral placebo capsules 200mg every eight hours for four weeks. One cycle is four weeks.
Patients will stay on medication until progression or reasons deem otherwise. The primary end-point is progression-free survival (PFS)— projected median PFS times for the two groups being eight and five months, respectively—and the secondary end-point is overall survival (OS). RECIST based on helical computed tomography (CT) scans will be used to determine disease progression, tumor response rate, and duration of response. This protocol has gained approval by the FDA under Special Protocol Assessment (SPA).
Clinical Study 7
A randomized, placebo-controlled phase Ib/IIa safety, tolerability, and efficacy study of oral PXD in combination with docetaxel versus docetaxel alone in patient with recurrent epithelial ovarian, fallopian tube, and primary peritoneal cancer is under way at Yale Cancer Center. A total of 60 patients will be enrolled and randomly allocated to either of the following two treatment arms: arm one—oral PXD 400mg every eight hours in combination with docetaxel IV 36mg/m2 weekly; arm two—placebo every eight hours in combination with docetaxel IV 36mg weekly. Patients will be treated for three cycles; one cycle consists of three weeks on and one week off.
The primary objective is to determine the safety and tolerability of combinational therapy of oral PXD and docetaxel in patients with recurrent ovarian cancer. The secondary objectives are to determine the effect of PXD on the toxicity of docetaxel using a weekly treatment regimen, to determine whether combinational therapy of PXD plus docetaxel is more efficacious than docetaxel therapy alone, to determine whether combinational therapy of PXD plus docetaxel affects blood levels of either drug, and to determine phenotypic differences in the tumor cells of ‘responders’ and ‘non-responders.’
Clincal Study 8
A phase II trial of PXD is being conducted at Yale Cancer Center and West Haven Veterans Administration of Connecticut in patients with castrate and non-castrate prostate cancer. A total of 60 eligible patients will be enrolled: 25 chemotherapy-naïvepatients with metastatic castrate disease are (hormonerefractory prostate cancer; Group A) and 35 patients with rising PSA after radical prostatectomy or radiotherapy who are non-castrate androgendependent (Group B). All patients will be treated with oral PXD 400mg every eight hours alone for 28 consecutive days (one cycle). Treatment outcome will be evaluated after three cycles (12 weeks) of PXD treatment (immediately prior to cycle four). The primary objective for this study is to determine the proportion of those treated with PXD who have a 50% post-therapy prostatespecific antigen (PSA) decline at 12 weeks.
Conclusion
The development of chemoresistance is a major limitation in the treatment of cancers. PXD is a novel biomodulator that has been shown to affect specific survival and death pathways. More importantly, it has been shown to reverse chemoresistance in vitro. In humans, it is well tolerated and has shown evidence of activity. The major challenge in the future is the development of an appropriate dosage form for PXD for its successful use in cancer treatment. ■
Phenoxodiol—A Chemosensitizer in the Midst of Cancer Chemoresistance
Article
References
- Alvero AB, O’Malley D, Brown D, et al., Molecular mechanism of phenoxodiol-induced apoptosis in ovarian carcinoma cells, Cancer, 2006;106:599–608.
- Constantinou AI, Husband A, Phenoxodiol (2H-1-benzopyran-7- 0,1,3-[4-hydroxyphenyl]) a novel isoflavone derivative, inhibits DNA topoisomerase II by stabilising the cleavable complex, Anticancer Res, 2002;22:2581–5.
- Constantinou AI, Mehta R, Husband A, Phenoxodiol, a novel isoflavone derivative, inhibits dimethylbenz[a]anthracene (DMBA)-induced mammary carcinogenesis in female Sprague–Dawley rats, Eur J Cancer, 2003;39:1012–18.
- Kamsteeg M, Rutherford T, Sapi E, et al., Phenoxodiol – an isoflavone analog – induces apoptosis in chemoresistant ovarian cancer cells, Oncogene, 2003;22:2611–20.
- Alvero AB, Brown D, Montagna M, et al., Phenoxodiol-Topotecan Co-Administration Exhibit Significant Antitumor Activity Without Major Adverse Side Effects, Cancer Biol Ther, 2007;6.
- Sapi E, Alvero AB, Chen W, et al., Resistance of ovarian carcinoma cells to docetaxel is XIAP dependent and reversible by phenoxodiol, Oncol Res, 2004;14:567–78.
- Aguero MF, Facchinetti MM, Sheleg Z, Senderowicz AM, Phenoxodiol, a novel isoflavone, induces G1 arrest by specific loss in cyclin-dependent kinase 2 activity by p53-independent induction of p21WAF1/CIP1, Cancer Res, 2005;65:3364–73.
- Gamble JR, Xia P, Hahn CN, et al., Phenoxodiol, an experimental anticancer drug, shows potent antiangiogenic properties in addition to its antitumour effects, Int J Cancer, 2006;118: 2412–20.
- Baudhuin LM, Cristina KL, Lu L, Xu Y, Akt activation induced by lysophosphatidic acid and sphingosine-1-phosphate requires both mitogen-activated protein kinase kinase and p38 mitogen-activated protein kinase and is cell-line specific, Mol Pharmacol, 2002;62:660–71.
- Kang KH, Kim WH, Choi KH, p21 promotes ceramide-induced apoptosis and antagonizes the antideath effect of Bcl-2 in human hepatocarcinoma cells, Exp Cell Res, 1999;253:403–12.
- De Luca T, Morre DM, Zhao H, Morre DJ, NAD+/NADH and/or CoQ/CoQH2 ratios from plasma membrane electron transport may determine ceramide and sphingosine-1-phosphate levels accompanying G1 arrest and apoptosis, Biofactors, 2005;25: 43–60.
- Scarlett DJ, Herst P, Tan A, et al., Mitochondrial gene-knockout (rho0) cells: a versatile model for exploring the secrets of trans-plasma membrane electron transport, Biofactors, 2004;20: 199–206.
- Morre DJ, Chueh PJ, Yagiz K, et al., ECTO-NOX target for the anticancer isoflavene phenoxodiol, Oncol Res, 2007;16:299–312.
Further Resources
Trending Topic
Worldwide, ovarian cancer is the seventh most common cancer and the eighth most common cause of cancer death in women.1 The GLOBOCAN study estimated there were 239,000 new cases in 2012 and 152,000 deaths due to this disease. There are nearly 600,000 women living within 5 years of an ovarian cancer diagnosis.1 In the EU, age-adjusted ovarian […]
Worldwide, ovarian cancer is the seventh most common cancer and the eighth most common cause of cancer death in women.1 The GLOBOCAN study estimated there were 239,000 new cases in 2012 and 152,000 deaths due to this disease. There are nearly 600,000 women living ...

Gynecological malignancies can be difficult to treat. Ovarian cancer carries with it the worst prognosis of all gynecological cancers. Approximately 75% of patients with epithelial ovarian cancer are diagnosed with advanced disease which is curable only in a minority of cases, ...

It is not by chance that this issue of European Oncology & Haematology is opened by an editorial from Suhag, dedicated to the oncological scenario in developing countries, warning that there is “little light at the end of the tunnel”. ...
Areader of 20 years ago could have hardly imagined many of the contents of this issue of European Oncology & Haematology. Twenty years ago, chronic myeloid leukemia (CML) was still treated with hydroxyurea, interferon and allogeneic stem cell transplantation, and multiple ...
Epithelial ovarian cancer has long been recognized as being highly sensitive to cytotoxic chemotherapy, with anticipated objective response to platinum-based chemotherapy of approximately 70-85%1. Furthermore, overall survival (OS) for women presenting with advanced disease has been documented to have improved ...
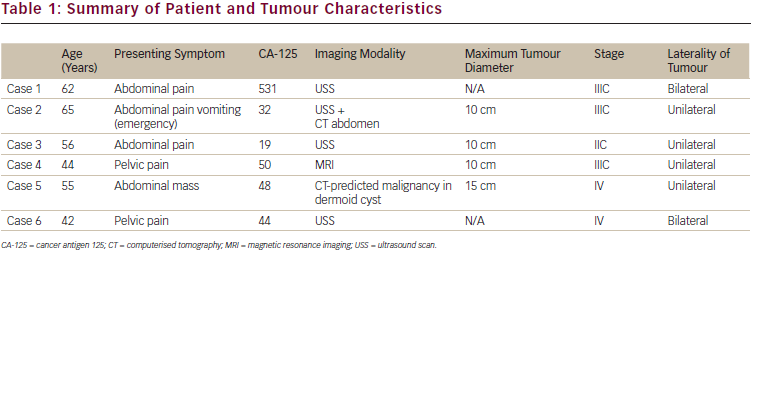
Mature cystic teratomas (also called dermoid cysts) account for about 30–45 % of all ovarian neoplasms and around 60 % of all benign tumours arising in the ovary.1 Malignant transformation of the various mature tissue components of a dermoid cyst is rare and the ...
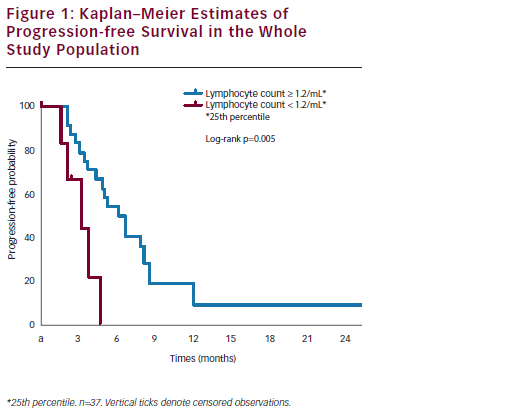
Ovarian cancer (OC) is the most lethal gynaecological cancer. In 2010, approximately 21,880 new cases and 13,850 deaths occurred in the US,1 while in the EU in 2004 the incidence of newly diagnosed cases was 42,700 with a mortality of 12/100,000 women/year.2 The current treatment ...
Axel Heidenreich is Chairman and Director of the Department of Urology at University Hospital, Aachen, Germany, whose main area of interest is prostate cancer. His recent scientific awards include the Paul-Mellin-Preis der Nordrhein Westfälischen Gesellschaft für Urologie (2010), the ...
Ovarian cancer is the most lethal gynaecological cancer. In the US, approximately 21,880 new cases were identified and 13,850 women died of the disease in 2010.1 In the European Union, the number of newly diagnosed cases was 42,700 in 2004 and the mortality rate is 12 ...
This issue of European Oncology highlights many of the challenges that we face in our fight against cancer in this diverse world. Clinical drug development has become complex, and independent academic research is in danger. Fortunately, these difficulties are not ...
Ovarian cancer is the fifth most common malignancy in women. In the US, approximately 22,000 new cases occur each year.1 In Germany, about 8,000 new cases are diagnosed each year. About 80% of patients suffer from advanced-stage disease (International Federation of Gynaecology and ...
Ovarian cancer is the fifth most common malignancy in women. In the US, approximately 22,000 new cases occur each year.1 In Germany, about 8,000 new cases are diagnosed each year. About 80% of patients suffer from advanced-stage disease (International Federation of Gynaecology andObstetrics [...
Log into your Touch Account
Keep track of your clinical interests and newsletter subscriptions.
Sign up with an Email
Or use a .
Register now for FREE access
Already registered? Login below.
