Rat sarcoma virus (RAS) proteins are a family of prototypical oncogenes frequently mutated in human cancers. Mutations in the RAS gene account for 19% of all pathogenic alterations and are the subject of extensive research in molecular and clinical oncology.1 The RAS family consists of three major isoforms, namely the Harvey rat sarcoma virus (HRAS), the neuroblastoma RAS viral oncogene homologue (NRAS) and the Kirsten rat sarcoma virus (KRAS). Amongst these, KRAS is the most frequently mutated of the RAS isoforms, with a remarkably high mutational prevalence in gastrointestinal (GI) malignancies, including 90% of pancreatic ductal adenocarcinomas (PDAC) and 50% of colorectal cancers (CRC).2 The high prevalence of KRAS aberrations in GI malignancies was crucial in promoting studies that identified KRAS’s central role as an oncogenic driver and a critical factor in resistance against cytotoxic chemotherapy.3 However, despite decades of research into its molecular configuration, efforts in targeting KRAS have been elusive for several reasons: the absence of deep pockets in the RAS protein, making it inaccessible to small-molecule inhibitors, the constant transition between two distinct conformational states with vastly different chemical behaviours and the high affinity for guanosine-5′-triphosphate (GTP), allowing the KRAS protein to function despite low GTP concentrations, have made the design of a targeted KRAS inhibitor particularly difficult.4
Nevertheless, advances in molecular biology and conformational biochemistry have profoundly changed our understanding of an anomalous mutant variant known as the Gly12Cys KRAS (KRAS G12C) mutant.5 The successful development of KRAS G12C inhibitors is attributed to the distinct presence of the switch II pocket that allows covalent inhibition of the cysteine residue, as well as the high GTPase activity of KRAS G12C that likely results in an attenuated duration of rapidly accelerated fibrosarcoma (RAF) kinase activation, which is not fully present in other KRAS-mutant types.6 Despite the ubiquity of KRAS mutations in solid tumours, the global prevalence of KRAS G12C mutations is relatively low in PDAC and CRC, accounting for only 1.3 and 3.1% of the total mutational burden, respectively (Table 1).9–11 Moreover, several studies in CRC and PDAC have found that KRAS mutations, particularly KRAS G12C, are associated with a poor response to cytotoxic chemotherapy.12 Indeed, patients with KRAS G12C-mutated CRC had a median overall survival (mOS) of 16.1 (95% confidence interval [CI], 13.0–19.0) months compared with 18.3 (95% CI, 17.2–19.3) months for KRAS non-G12C-mutated tumours and 19.2 (18.5–19.8) months for the mCRC overall cohort .13,14 In PDAC, mOS was 16.7 months in KRAS G12C-mutated tumours versus 24.9 months in KRAS non-G12C-mutated tumours.15 This review aims to summarize the pleiotropic functions of the KRAS gene, highlight the unique features of the KRAS G12C protein concerning its biological role and targeted treatments, describe the main mechanisms of resistance of currently approved drugs and finally review the results from published clinical trials testing KRAS G12C inhibitors, as well as KRAS G12C downstream and upstream protein inhibitors, with a focus on the most promising ongoing trials.
Table 1: Prevalence and distribution of Kirsten rat sarcoma virus G12 mutations across gastrointestinal cancer types7,8
|
|
Colorectal (%) |
Pancreas (%) |
Gastric/Esophageal (%) |
Gallbladder/cholangiocarcinoma (%) |
|
KRAS G12C |
7.0 |
1.3 |
5.9/7.6 |
0.5/1.2 |
|
KRAS G12D |
29.9 |
41.8 |
27.0/33.0 |
33.6 |
|
KRAS G12V |
20.0 |
31.6 |
18.0/13.0 |
27.2 |
|
KRAS G12R |
1.1 |
16.1 |
3.9/0.8 |
9.6 |
KRAS = Kirsten rat sarcoma virus.
Kirsten rat sarcoma virus structure and effects of activating Kirsten rat sarcoma virus mutations
RAS proteins are small, membrane-bound guanine nucleotide-binding proteins. The major RAS isoforms are encoded by three genes, HRAS, NRAS and KRAS, producing four proteins, HRAS, NRAS, KRAS4A and KRAS4B, from a KRAS splice variant. The amino-terminal residues 1–165 of these proteins share 92–98% sequence similarity, and the remaining 23–24 carboxy-terminal residues diverge significantly. The structural domain of the RAS protein consists of the first 166–168 residues, forming the G domain, a structure featuring a mix of six-stranded β-sheet and five-α-helix fold, typical of α,β-nucleotide-binding proteins. Four main regions border the nucleotide-binding pocket: the phosphate-binding loop (P-loop, residues 10–17), switch I (residues 30–38), switch II (residues 60–76) and the base-binding loops (residues 116–120 and 145–147).16 Similar to its RAS family counterparts, NRAS and HRAS, KRAS mediates downstream signalling via the activation of the RAF protein/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinases (ERK) cascade, the phosphoinositide-3-kinase complex/mammalian target of rapamycin (PI3K/mTOR) axis and ultimately the RAS-related protein/nuclear factor-κB (NF-κB) pathway, driving tumour onset, progression and distant spread.17–19 KRAS constantly transitions between an inactive ‘off’ state when bound to guanosine diphosphate (GDP) and an active ‘on’ state when bound to GTP, a process facilitated by guanine nucleotide exchange factors (GEFs) such as Son of Sevenless (SOS) 1 and 2. In the GTP-bound state, threonine-35 (in the switch I region) and glycine-60 (in the switch II region) stabilize the active conformations of the switch I and switch II regions by anchoring to the γ-phosphate of GTP. Upon phosphate release during GTP hydrolysis, switch I and switch II recoil back into their inactive GDP conformations, such that these regions ultimately regulate all known nucleotide-dependent interactions between RAS and its binding partners.16 The exchange of GDP for GTP results in the activation and dimerization of RAS proteins, ultimately leading to the propagation of signal transduction cascades. This transition is mainly regulated by a variety of upstream receptor tyrosine kinases (RTKs) such as the epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), human epidermal growth factor receptor 3 and fibroblast growth factor receptors (FGFRs).20 Most KRAS-activating mutations trigger the conversion of KRAS-GDP into KRAS-GTP, further enhanced by inhibiting GTPase activity that locks KRAS in its ‘on’ state, leading to the constitutive activation of downstream signalling pathways (Figure 1).
Figure 1: Schematic representation of Kirsten rat sarcoma virus G12C signalling pathways and regulatory mechanisms
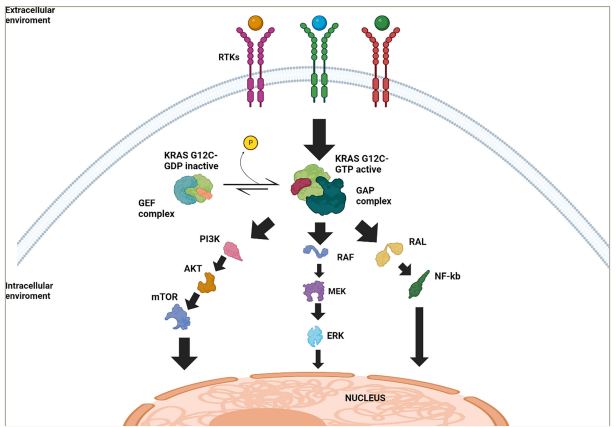
Simplified illustration of the signalling pathways downstream of KRAS G12C in its active and inactive states. Black arrows indicate upregulation or activation. At the cell membrane, RTKs initiate the activation of KRAS. In the inactive state, KRAS G12C is bound to GDP and undergoes activation through interaction with GEFs, which facilitate the exchange of GDP for GTP. Once bound to GTP, KRAS G12C transitions to its active state and promotes the activation of multiple downstream signalling pathways (RAF/MEK/ERK, PI3K/AKT/mTOR pathway and RAL/NF-κB). The GAPs promote the hydrolysis of GTP to GDP, inactivating KRAS G12C and returning it to the inactive GDP-bound state. These signalling pathways ultimately converge at the nucleus, regulating cellular processes such as proliferation, survival and tumour progression.
AKT = protein kinase B; ERK = extracellular signal-regulated kinase; GAP = GTPase-activating protein; GDP = guanosine diphosphate; GEF = guanine nucleotide exchange factors;
GTP = guanosine-5‘-triphosphate; KRAS = Kirsten rat sarcoma virus; MEK = mitogen-activated protein kinase kinase; mTOR = mammalian target of rapamycin; NF-kB = nuclear
factor-κB; PI3K = phosphoinositide3-kinase complex; RAF = rapidly accelerated fibrosarcoma; RAL = RAS-related protein; RTKs = receptor tyrosine kinase.
Evolution of Kirsten rat sarcoma virus-targeting strategies
Targeting KRAS has been an important research challenge during the last 30 years when it was considered ‘undruggable’. This challenge stemmed in part from the structure of KRAS, a small protein with a relatively smooth surface that lacks well-defined hydrophobic pockets (except for its GTP/GDP-binding pocket) suitable for small-molecule inhibitor binding, as well as from KRAS’s ability to bind GTP with picomolar affinity in an environment with high intracellular GTP concentrations.16,21–24 In addition, each KRAS mutation alters the structure of the GTP-binding pocket differently, which further complicates the design of an inhibitor that is effective against multiple KRAS alterations.25–27 Strategies therefore focused on indirectly targeting KRAS by inhibiting downstream signalling effectors such as PI3K and MEK, modifying the epigenetic environment with telomerase inhibitors, and RNA interference or promoting synthetic lethality using cyclin-dependent kinase inhibitors, with limited tolerability and efficacy.28–31 In the last decade, molecular studies of KRAS-mutated proteins highlighted the unique structure of the aberrant variant KRAS G12C, resulting from a missense mutation, causing the substitution of glycine with cysteine at codon 12 in exon 2. The presence of cysteine causes a steric block that prevents the arginine finger of GTPase-activating proteins from binding to the GTPase site of RAS, inhibiting the hydrolysis of GTP to GDP, thus maintaining KRAS in a constitutively active state.32 However, a distinguishing feature of the KRAS G12C protein, discovered by Shokat’s laboratory in 2013, is the presence of an allosteric pocket below the switch II region of the mutated cysteine residue, called the switch II pocket, which is susceptible to inhibition through covalent binding.20 Unlike other KRAS mutations, KRAS G12C does not significantly alter the intrinsic GTPase activity, allowing KRAS to remain in the GDP-bound or inactive state for a longer period.33 These biochemical and functional properties paved the way for the development of the first KRAS G12C-targeted therapies: sotorasib and adagrasib. Both drugs irreversibly bind the cysteine residue in the switch II pocket of KRAS G12C, locking it in its inactive GDP-bound state and inhibiting downstream signalling.16,34 While adagrasib has high selectivity for KRAS G12C, sotorasib inhibits NRAS G12C more potently compared with KRAS G12C or HRAS G12C. According to structural and reciprocal mutagenesis studies, differences in isoform-specific binding are mediated only by histidine-95 in KRAS and leucine-95 in NRAS.35 A single patient with NRAS G12C CRC was reported to have had a marked tumour response after being treated with sotorasib and panitumumab, suggesting that sotorasib can be clinically effective in NRAS G12C-mutated tumours.35 Regardless, the clinical application of single-agent sotorasib was studied in 2021 to treat advanced KRAS G12C-mutated non-small-cell lung cancer (NSCLC) in the CodeBreaK 100 trial (A Phase 1/2, Open-label Study Evaluating the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Efficacy of Sotorasib [AMG 510] Monotherapy in Subjects With Advanced Solid Tumors With KRAS p.G12C Mutation and Sotorasib [AMG 510] Combination Therapy in Subjects With Advanced NSCLC With KRAS p.G12C Mutation; ClinicalTrials.gov identifier: NCT03600883).36 This phase II trial enrolled 126 patients (pts) with previously treated KRAS G12C-mutated NSCLC who received sotorasib. The overall response rate (ORR) was 37.1% (95% CI, 28.6–46.2), disease control rate (DCR) was 80.6% (95% CI, 72.6–87.2), median progression-free survival (mPFS) was 6.8 months (95% CI, 5.1–8.2) and mOS was 12.5 months.36 The CodeBreaK 100 trial has been considered a landmark trial based on these results, and sotorasib was granted accelerated approval by the US Food and Drug Administration (FDA) in May 2021 for the treatment of locally advanced or metastatic KRAS G12C-mutated NSCLC pts who have failed one or more systemic therapies.37. Comparable findings were observed with adagrasib monotherapy in a similar population, prompting its approval by the US FDA in December 2022.38 Sotorasib and adagrasib have also shown encouraging but varied results in GI cancers. The initial findings from the CodeBreaK 100 trial included a cohort of 12 pts with KRAS G12C-mutated PDAC who received sotorasib monotherapy, amongst whom one achieved a partial response (PR).39 When the trial was expanded to include 38 pts with KRAS G12C-mutated PDAC, mPFS was 4.0 months and mOS was 6.9 months. In addition, 21% of pts achieved a PR, with a DCR of 84%.40 In the phase I–II KRYSTAL-1 trial (ClinicalTrials.gov identifier: NCT03785249), adagrasib monotherapy demonstrated comparable outcomes to sotorasib: for 21 pts with metastatic PDAC receiving adagrasib, mPFS was 5.4 months (95% CI, 3.9–8.2) and mOS was 8.0 months (95% CI, 5.2–11.8). ORR was 33% and DCR was 49%.41 So far, both adagrasib and sotorasib have been adopted as single-agent therapeutic options by the National Comprehensive Cancer Network guidelines for advanced KRAS G12C-mutated PDAC and biliary tract cancers (BTC).41 While single-agent KRAS G12C inhibitors have shown promising results in PDAC, their efficacy appears less pronounced in CRC. On the other hand, improved results were observed testing sotorasib and adagrasib in combination with EGFR inhibitors. The analysis of CodeBreaK 101, a phase Ib study of pts with chemorefractory KRAS G12C-mutated CRC receiving sotorasib alone or in combination with EGFR inhibitors, showed an ORR of 30% with sotorasib–panitumumab compared with 9.7% with sotorasib monotherapy.42,43 Subsequently, the CodeBreaK 300 trial evaluated two doses of sotorasib (960 mg once daily and 240 mg once daily) in combination with panitumumab versus standard-of-care trifluridine/tipiracil in chemorefractory metastatic CRC (mCRC) and showed an mPFS of 5.6 and 3.9 months in the 960 mg sotorasib–panitumumab group and 240 mg in the sotorasib–panitumumab group compared with 2.2 months in the standard-care group. ORR was 26.4, 5.7 and 0% for the 960 mg sotorasib–panitumumab, 240 mg sotorasib–panitumumab and standard-care groups, respectively.42 Of note, the phase I–II KRYSTAL-1 trial assessed adagrasib ± cetuximab in the same setting, showing an ORR of 34% (95% CI, 24.6–44.5) versus 21.4% (95% CI, 10.3–36.8) in favour of the combination treatment with a similar DCR (85.1 versus 86.2%) between the two arms. The combination confirmed its superiority in all the other efficacy outcomes, with an mPFS of 6.9 months (95% CI, 5.7–7.4) versus 4.1 months (95% CI, 2.8–6.5). mOS was 15.0 months (95% CI, 11.8–18.8) versus 12.2 months (95% CI, 8.1–15.2). Interestingly, the combination was more tolerated with 27% of grade 3–4 treatment-related adverse events (TRAEs) versus 34%, respectively.44 Fuelled by these positive results, the KRYSTAL-10 (ClinicalTrials.gov identifier: NCT04793958), a global, open-label, randomized phase III study, evaluates adagrasib and cetuximab against chemotherapy in the same patient cohort.45 On the other hand, the use of KRAS G12C inhibitors remains limited in other GI cancers. The prevalence of KRAS G12C varies across GI malignancies: it is limited in BTC, accounting for only 1% of mutations, and is slightly more prevalent in appendiceal cancers (3–4%).10 A cohort of KRYSTAL-1 focusing on non-CRC GI cancers evaluated 57 pts, amongst whom 12 had BTC and 16 had other GI tumours (nine pts had appendiceal cancer, four had gastro-oesophageal junction/oesophageal cancer and three had small bowel cancer). Updated results published in May 2023 demonstrated an ORR of 35.1%, a DCR of 86.0% and a median duration of response of 5.3 months (95% CI, 2.8–7.3). mPFS was 7.4 months (95% CI, 5.3–8.6) and mOS was 14.0 months (95% CI, 8.5–18.6). Interestingly, in pts with BTC, ORR was 41.7%, DCR was 91.7%, mPFS was 8.6 months (95% CI, 2.7–11.3) and mOS was 15.1 months (95% CI, 8.6–not estimable).41
Emergence of resistance
Despite the initial clinical responses achieved with KRAS G12C inhibitors, treatment failure eventually occurs due to the emergence of resistance. Resistance mechanisms can be divided into genetic when a mutation is identified in genes involved in RAS-dependent molecular pathways and adaptive when reactivation of upstream proteins occurs. According to paired plasma sample analyses, genetically acquired mutations have been detected in 90% of mCRC pts treated with the new KRAS G12C inhibitor divarasib with or without cetuximab.46 The most common on- and off-target alterations detected include new activating KRAS mutations, upstream RTKs hyperactive alterations (EGFR and FGFRs) and aberrations associated with downstream proteins (PIK3CA, RAF and MEK). Similarly, pts treated with adagrasib with or without cetuximab, for whom genomic-acquired mechanisms of resistance were detectable in more than 70% of pts, the newly acquired mutations affected both downstream signalling proteins, such as mitogen-activated protein kinase (MAPK) and PI3K, and upstream RTKs.47 Adaptive resistance most commonly manifests through the reactivation of upstream RTKs in response to KRAS G12C inhibition, limiting the efficacy of targeted drug therapies.48 Mutant KRAS G12C suppresses the activation of upstream RTKs and other wild-type RAS isoforms through ERK-mediated feedback inhibition. Therefore, using an agent stabilizing KRAS G12C protein in its ‘off’ state relieves this suppression with a consequent upregulation of RTKs, ultimately leading to the activation of wild-type RAS isoforms. This circuit was demonstrated in two studies by Ryan et al., where treatment with KRAS G12C inhibitors resulted in a rapid rebound increase in MAPK signalling as a result of the increased expression of multiple upstream RTKs, including EGFR, HER2 and FGFR.49,50 Uncovering these resistance mechanisms has fuelled the development of novel targeted therapeutic agents, which are under clinical investigation as both single-agent and combined strategies with other agents. Several promising therapies are currently undergoing preclinical or early-phase trials within multicohort studies, with the most relevant highlighted in Table 2.p28,36,51–65
Table 2: Comprehensive list of new Kirsten rat sarcoma virus G12C ‘off’ inhibitors and their treatment-related adverse events28,36,51–65
|
Agent |
Pharmaceutical company |
Clinical trial and identifier |
Incidence of grade ≥3 TRAEs |
Most commonly reported grade ≥3 TRAEs |
|
LY353798251 |
Eli Lilly and Company |
Phase I (NCT04956640) |
NA |
Diarrhoea, constipation, fatigue, peripheral oedema, nausea and neutropenia |
|
GDC-603628 |
Genentech |
Phase I (NCT04449874) |
12% |
Increased ALT, increased AST, nausea, vomiting and fatigue |
|
D-155352 |
InventisBio |
Phase I/II (NCT04585035) |
22% |
Increased AST and ALT, diarrhoea, hypertension, hypokalaemia and nausea |
|
HBI-243853 |
Huyabio International |
Phase I (NCT05485974) |
Pending |
Pending |
|
JDQ44354 |
Novartis |
Phase Ib/II (NCT04699188) |
7.1% |
Neutropenia, increased ALT and AST and myalgia |
|
JAB-2182255 |
Jacobio Pharma |
Phase I/II (NCT05009329) |
0% |
NA |
|
HS-1037036 |
Jiangsu Hansoh Pharmaceutical Company |
Phase I/II (NCT05367778) |
27.3% |
Increased AST and ALT, anaemia, diarrhoea, weight gain, decreased appetite, hypoproteinaemia, nausea, fatigue and rash |
|
IBI-351 (GFH925)56 |
Innovent Biologics Inc. |
Phase I/II (NCT05005234) |
20% |
Anaemia, leukopaenia, increased ALT and pruritus |
|
BI-182391157 |
Boehringer Ingelheim |
Phase I (NCT04973163) |
30% |
Nausea, diarrhoea, vomiting, fatigue and decreased appetite |
|
JNJ-7469915758 |
Johnson & Johnson |
Phase I (NCT04006301) |
55% |
Increased blood CPK |
|
GFH92556 |
GenFleet |
Pooled analysis of two phase I studies (NCT05005234 and NCT05497336) |
20% |
Anaemia, decreased white blood cell count, increased ALT and pruritus |
|
YL-1529359 |
Shanghai Yingli |
Phase I (NCT05173805) |
NA |
NA |
|
BPI042128660 |
Belta |
Phase I (NCT05315180) |
Pending |
Pending |
|
GH3561 |
Suzhou Genhouse Bio |
Phase I (NCT05010694) |
Pending |
Pending |
|
GEC25562 |
GenEros Biopharma |
Phase I |
6.7% |
Diarrhoea, increased ALT, rash and anaemia |
|
MK-108463 |
Merck |
Phase I (NCT05067283) |
Pending |
Pending |
|
D3S-00164 |
D3 Bio |
Phase I |
14.6% |
NA |
|
HBI-243866 |
Huyabio |
Phase I (NCT05485974) |
Pending |
Pending |
|
SY-593365 |
Shouyao Holdings |
Phase I (NCT06006793) |
Pending |
Pending |
ALT = alanine aminotransferase; AST = aspartate aminotransferase; CPK = blood creatinine phosphokinase; NA = not available; TRAEs = treatment-related adverse events.
New direct inhibitors of Kirsten rat sarcoma virus G12C
Since the FDA approved adagrasib and sotorasib, several KRAS G12C inhibitors with different safety profiles are entering clinical trials. Divarasib is a KRAS G12C inhibitor that binds to the KRAS G12C protein in its ‘off’ state. It is, however, up to 50 times more selective and 5–20 times more potent than sotorasib and adagrasib according to in vitro studies.33 Divarasib monotherapy was evaluated in a phase I trial enrolling 137 pts, 55 of whom had mCRC.32 No dose-limiting toxicity occurred across concentrations ranging from 50 to 400 mg, and grade ≥3 TRAEs were observed in 12% of pts, leading to treatment discontinuation in 3% of cases. Amongst patients in the CRC cohort treated with divarasib 400 mg daily, 35.9% had a confirmed response (complete response [CR] or PR) and an mPFS of 6.9 months. More recently, the combination of divarasib with cetuximab achieved an improved ORR of 62.5% (95% CI, 40.6–81.2%) and an mPFS of 8.1 months (95% CI, 5.5–12.3) in pts with mCRC enrolled in a phase Ib trial (A Phase Ia/Ib Dose-escalation and Dose-expansion Study Evaluating the Safety, Pharmacokinetics, and Activity of GDC-6036 as a Single Agent and in Combination with Other Anti-cancer Therapies in Patients with Advanced or Metastatic Solid Tumors with a KRAS G12C Mutation; ClinicalTrials.gov identifier: NCT04449874).67 In a limited PDAC cohort containing seven pts receiving divarasib, three achieved PR, while four had stable disease (SD). Similarly, amongst seven pts with BTC, one patient had a PR, four had SD and one had progressive disease (PD).46 Therefore, further assessment of divarasib in PDAC and BTC is warranted in more extensive randomized trials. Divarasib is under evaluation in two phase I basket trials (ClinicalTrials.gov identifiers: NCT04449874 and NCT04929223), exploring its efficacy in combination with cetuximab with or without cytotoxic chemotherapy.68,69 Garsorasib is another orally bioavailable small-molecule inhibitor of KRAS G12C shown to be highly potent in vivo using cell line-derived and patient-derived xenograft tumour models, as well as in PDAC and CRC in vitro cell lines. In CRC patient-derived xenografts models, single-agent demonstrated tumour growth inhibition ranging from 60.9 to 105.7%, with three out of nine models showing tumour regression.70 Combining garsorasib with other anti-cancer agents, such as MEK inhibitors, tyrosine phosphatase-2 (Src Homologous Protein 2 [SHP2]) inhibitors and chemotherapy, increased its anti-tumoural activity and enhanced tumour regression.71 The efficacy of garsorasib was evaluated in combination with cetuximab in an ongoing phase II trial (ClinicalTrials.gov identifier: NCT04585035) in pts with refractory mCRC. Preliminary results demonstrated an ORR of 45% and a DCR of 95%. However, data on overall survival are still pending.70–72 Similarly, amongst 10 pts with advanced PDAC who received garsorasib, ORR was 50%, including one CR, DCR was 80% and mPFS was 8.54 months.73 It is essential to mention that in this trial, only 40% of pts received two or more lines of therapy.
Son of Sevenless 1 and Src Homologous Protein 2 inhibitors
Son of Sevenless 1 (SOS1) and SHP2 are signalling intermediates activated by RTKs that act as central nodes in the RAS signalling pathway. SOS1 is a GEF protein that binds inactive KRAS–GDP complex and mediates GTP exchange. SHP2, an adaptive phosphatase, links directly to SOS1, further facilitating GTP exchange. Therefore, inhibiting both SOS1 and SHP2 blocks RTK-positive molecular signalling to KRAS, maintaining it in its inactive GDP state. Published data demonstrated that co-inhibition of SHP2 and KRAS G12C overturns feedback reactivation across different RTKs and that combined KRAS G12C–SHP2 inhibition maintains RAS pathway suppression with improved efficacy both in vitro and in vivo.49,50 Since new activating RTK alterations were also commonly detected in liquid biopsies at the time of radiological treatment progression, approaches that target KRAS activation via SOS1 or SHP2 inhibitors represent a compelling avenue for intervention. The combination of glecirasib with JAB-3312, an SHP2 inhibitor, in pts with NSCLC with no prior exposure to KRAS G12C inhibitors demonstrated an ORR of 50% and a DCR of 100%, albeit with increased toxicity.74 RMC-4630 is another SHP2 inhibitor being examined with sotorasib in the CodeBreaK 101 trial.75 In a cohort of six pts, five (83%) achieved disease control; however, no responses were observed.
Downstream mitogen-activated protein kinase blockade
As mentioned previously, adaptive and acquired resistance mechanisms may involve upregulation of the RTK/RAS/MAPK downstream signalling pathway, hence the rationale behind using RAS downstream inhibitors. A well-described example is the upregulation of EGFR that occurs with B-Raf murine sarcoma viral oncogene homologue B (BRAF) inhibition in BRAF-mutated CRC, necessitating the addition of EGFR inhibitors to counteract drug resistance. Another example is the compensatory activation of the mTOR pathway, which has been documented in KRAS G12C inhibitor-resistant CRC cell lines.76 Currently, mTOR inhibitors such as everolimus are being explored in combination with sotorasib in NSCLC, while relevant phase I basket trials are investigating combinations such as adagrasib with nab-sirolimus (ClinicalTrials.gov identifier: NCT05840510) and divarasib with the PI3K inhibitor inavolisib (ClinicalTrials.gov identifier: NCT04449874) in pts with various solid tumours, including CRC.68,77 Moreover, the addition of MEK inhibitors to G12C inhibitors is being tested in the CodeBreaK 101 study as a new alternative, based on preclinical studies showing that sotorasib and trametinib (MEK inhibitor) combination has a synergistic anti-tumour effect on NSCLC tumour cell lines in vitro compared with either of the single agents alone.78 In this context, sotorasib and trametinib (MEK inhibitor) were evaluated in 36 pts, 18 of whom have KRAS G12C-mutant CRC, achieving a remarkable DCR of 86%.79 It is worth noting that some pts had prior exposure to KRAS G12C inhibitors, and upon receiving the maximum tolerated dose of 2 mg trametinib and 960 mg sotorasib, all pts had radiological SD. These results, albeit reported in a limited number of pts, indicate a clinically significant therapeutic benefit associated with the addition of trametinib to sotorasib. Of note, this trial reported 34% of pts with grade ≥3 toxicity, leading to therapy discontinuation in 24% of cases.
Combination with immunotherapy
The tumour microenvironment (TME) in PDAC is a complex and dynamic cell network involving immune, stromal and cancer cells, facilitating tumour progression and response to therapy. KRAS mutations have been widely shown to foster anti-inflammatory and pro-inflammatory effects on the TME. Some studies underpinned that KRAS mutations promote an immunosuppressive TME through several mechanisms such as tumour cell expression of inhibitory cytokines interleukin (IL)-10 and transforming growth factor-β, recruitment of myeloid-derived suppressor cells, regulatory T-cell (T-reg cell) activation and suppression of cluster of differentiation 8 (CD8) T-cell activity.21,80 Other studies suggest that KRAS mutations could also interfere with the secretion of pro-inflammatory cytokines, such as intercellular adhesion molecule-1, tumour necrosis factor-α, IL-1β, IL-6 and IL-18, through the induction of NF-κB.81–83 On the other hand, adagrasib and sotorasib were found to induce a pro-inflammatory microenvironment and modulate the TME by recruiting macrophages, dendritic cells and CD8 T-cells, promoting anti-tumour immune response in NSCLC. These findings suggest a potential synergistic interaction between these agents and immune checkpoint inhibitors (ICIs).84,85 However, conflicting data have emerged regarding the efficacy and tolerability of these combined regimens. Early data from the CodeBreaK 101 study, in which pts with NSCLC received sotorasib plus pembrolizumab or atezolizumab, showed a high incidence of liver toxicity, with an ORR of 29% and a DCR of 83%.86 However, results from the KRYSTAL-7 (ClinicalTrials.gov identifier: NCT04613596) study combining adagrasib and pembrolizumab in pts with NSCLC who had programmed death-ligand 1 expression >50% demonstrated an ORR of 63% and a DCR of 84%, with limited high-grade toxicity (10%).87 The combination of KRAS G12C inhibitors with ICIs and other target therapies is currently being assessed in several clinical trials across other KRAS G12C-mutant solid tumours, PDAC and CRC (Table 3).40–42,44–46,51,56–58,67,69,72,74,75,79,88–107
Table 3: Summary of clinical trials testing Kirsten rat sarcoma virus G12C inhibitors as monotherapy or in combination with other agents in gastrointestinal cancers40–42,44–46,51,56–58,67,69,72,74,75,79,88–107
|
Target |
Drugs |
Trial name and identifier |
Phase |
GI malignancy |
Number of pts in the trial, if applicable |
Clinical efficacy |
|
KRAS G12C ‘off’ inhibitors |
Adagrasib41 |
KRYSTAL-1 (NCT03785249) |
I/II |
PDAC |
21 |
ORR: 33% DCR: 49% mPFS: 5.4 months mOS: 8.0 months |
|
CRC |
43 |
ORR: 19% DCR: 86% mPFS: 5.6 months mOS: 19.8 months |
||||
|
BTC |
12 |
ORR: 42% DCR: 92% |
||||
|
Appendiceal adenocarcinoma |
7 |
ORR: 0% DCR: 86% |
||||
|
GEA |
3 |
ORR: 33% DCR: 66% |
||||
|
Adagrasib+cetuximab44,45 |
NCT05634525 |
I |
PDAC |
Pending |
Pending |
|
|
KRYSTAL-1 (NCT03785249) |
I/II |
CRC |
28 |
ORR: 46% DCR: 100% mPFS: 6.9 months mOS: 13.4 months |
||
|
Adagrasib+cetuximab+irinotecan88 |
NCT05722327 |
I |
CRC |
Pending |
Pending |
|
|
Adagrasib+cetuximab versus chemotherapy89 |
KRYSTAL-10 (NCT04793958) |
III |
CRC, second line |
Pending |
Pending |
|
|
Adagrasib+TNO15590 |
KRYSTAL-2 (NCT04330664) |
I/II |
CRC |
Pending |
Pending |
|
|
Adagrasib+BI 170196391 |
KRYSTAL-14 (NCT04975256) |
I |
CRC |
Pending |
Pending |
|
|
Adagrasib+MRTX090292 |
NCT05578092 |
I/II |
NA |
Pending |
Pending |
|
|
Adagrasib+durvalumab93 |
NCT05848843 |
I |
CRC |
Pending |
Pending |
|
|
Adagrasib+INCB09928094 |
NCT06039384 |
I |
CRC |
Pending |
Pending |
|
|
Sotorasib40,42 |
CodeBreaK 100 (NCT03600883) |
I/II |
PDAC |
38 |
ORR: 21% DCR: 84% mPFS: 4.0 months mOS: 6.9 months |
|
|
CRC |
62 |
ORR = 9.7% DCR = 82% |
||||
|
Sotorasib+panitumumab95 |
CodeBreaK 101 (NCT04185883) |
I/II |
CRC |
40 |
ORR: 30% DCR: 93% mPFS: 5.7 months |
|
|
Sotorasib+panitumumab+FOLFIRI96 |
CodeBreaK 101 (NCT04185883) |
I/II |
CRC |
31 |
ORR: 58.1% |
|
|
Sotorasib+trametinib79 |
CodeBreaK 101 (NCT04185883) |
I/II |
CRC |
18 |
ORR: 11.1% DCR: 83.3% |
|
|
Sotorasib+panitumumab versus standard of care97 |
CodeBreaK 300 (NCT05198934) |
III |
CRC |
|
ORR: 26.4 versus 0% (SOC) DCR: 71.7 versus 46.3% (SOC) mPFS: 5.6 versus 2.2 months (SOC) |
|
|
Sotorasib+BI 170196398 |
CodeBreaK 101 (NCT04185883) |
I/II |
NA |
Pending |
Pending |
|
|
Sotorasib+RMC-463075 |
CodeBreaK 101 (NCT04185883) |
I/II |
CRC |
6 |
ORR: 0% DCR: 83.3% |
|
|
Sotorasib+BBP-39899 |
NCT05480865 |
I/II |
NA |
Pending |
Pending |
|
|
BI1823911 monotherapy+BI 170196357 |
NCT04973163 |
I |
NA |
Pending |
Pending |
|
|
Divarasib46 |
NCT04449874 |
I |
CRC |
55 |
ORR: 29% DCR: 85% mPFS: 5.6 months |
|
|
PDAC |
7 |
ORR: 43% DCR: 100% |
||||
|
BTC |
5 |
ORR: 0% DCR: 80% |
||||
|
Divarasib+cetuximab67 |
NCT04449874 |
I |
CRC |
29 |
ORR: 62.5% |
|
|
Divarasib+cetuximab+FOLFOX or FOLFIRI69 |
INTRINSIC (NCT04929223) |
I |
CRC |
Pending |
Pending |
|
|
Fulzerasib56 |
Pooled analysis of NCT05005234 and NCT05497336 |
I |
CRC |
45 |
ORR 43.8% DCR 87.5% |
|
|
Garsorasib72 |
NCT04585035 |
I/II |
CRC |
24 |
ORR: 21% DCR 95.8% mPFS: 7.6 months |
|
|
Garsorasib72 |
NCT04585035 |
I/II |
PDAC |
10 |
ORR: 50% DCR: 80% mPFS: 8.5 months |
|
|
Garsorasib+cetuximab72 |
NCT04585035 |
I/II |
CRC |
40 |
ORR: 45% DCR: 95% mPFS 7.6 months |
|
|
|
Glecirasib101 |
NCT05009329 and NCT05002270 |
I/II |
PDAC, biliary tract, gastric, small bowel, appendiceal, hepatocellular, peritoneal |
PDAC = 28 Others = 19 |
ORR: 46.4% DCR: 96.4% mPFS: 5.5 months |
|
Glecirasib+cetuximab102 |
NCT05002270 and NCT05194995 |
I/II |
CRC, small bowel and appendiceal adenocarcinoma |
43 |
ORR: 62.8% DCR: 93% mPFS: Not reached |
|
|
Glecirasib103 |
NCT06008288 |
II |
PDAC |
Pending |
Pending |
|
|
Glecirasib+JAB-331274 |
NCT05288205 |
I/II |
CRC and PDAC |
Pending |
Pending |
|
|
JDQ443 monotherapy+TNO155 or +tislelizumab104 |
KontRASt-01 (NCT04699188) |
I/II |
CRC |
Pending |
Pending |
|
|
|
JDQ443+trametinib+ribociclib or + cetuximab105 |
KontRASt-03 (NCT05358249) |
I/II |
CRC |
Pending |
Pending |
|
JNJ-7469915758 |
NCT04006301 |
I |
CRC |
|
Withdrawn from market |
|
|
LY353798251 |
LOXO-RAS-20001 (NCT04956640) |
I/II |
PDAC |
12 |
ORR: 42% DCR: 92% |
|
|
CRC |
20 |
ORR: 10% DCR: 90% |
||||
|
Others |
21 |
ORR: 52% DCR: 95% |
||||
|
LY3537982+cetuximab51 |
LOXO-RAS-20001 (NCT04956640) |
I/II |
CRC |
11 |
ORR: 45% DCR: 100% |
|
|
KRAS G12C ‘on’ inhibitors |
BBO-8520106 |
Preclinical |
|
|
|
|
|
|
RMC-6291107 |
NCT05462717 |
I |
CRC |
20 |
ORR = 40% DCR = 80% |
BTC = biliary tract cancer; CRC = colorectal cancer; DCR = disease control rate; FOLFIRI = leucovorin calcium (folinic acid)–fluorouracil–irinotecan hydrochloride; FOLFOX = leucovorin calcium (folinic acid)–fluorouracil–oxaliplatin; GEA = gastro-oesophageal cancer; GI = gastrointestinal;KRAS = Kirsten rat sarcoma virus; mOS = median overall survival; mPFS = median progression-free survival; NA = not available; ORR = overall response rate; PDAC = pancreatic ductal adenocarcinoma;pts = patients; SOC = standard of care.
Conclusion
Since the first demonstration of sotorasib efficacy with the published results from the CodeBreaK 100 trial in 2019, the current landscape of RAS targeting strategy has shifted from undruggable to druggable. Fuelled by the successful implementation of adagrasib and sotorasib as standard treatment options in PDAC, and recently in BTC and CRC, more than a dozen small-molecule inhibitors targeting KRAS G12C and related upstream and downstream molecules have emerged. They are actively investigating, with many of them already advanced to clinical trials for evaluation in GI cancers. However, despite the crucial development and relative success of KRAS G12C inhibitors in treating GI cancers, clinical research must still prioritize strategies to overcome primary and acquired resistance, primarily through vertical inhibition with RTK/RAS/MAPK pathway inhibitors and combination with immunotherapy or standard chemotherapy. In addition, more data about toxicity management and quality of life are awaited to ensure the safety of new drugs and combinations under evaluation. Other important considerations will be integrating KRAS G12C inhibitors in earlier phases of treatment, such as in the neoadjuvant and adjuvant settings. These strategies may expand the population of pts with GI cancers, benefitting from the inhibition of KRAS G12C. Undoubtedly, we are in a new and promising era of cancer treatment, pushing the boundaries of KRAS target therapy.