Waldenström macroglobulinemia (WM) is an indolent lymphoma characterized by the presence of a circulating monoclonal immunoglobulin M (IgM) protein of any size and lymphoplasmacytic infiltrate in the bone marrow.1 It lies within a continuous spectrum of disorders associated with a monoclonal IgM protein in the serum, with the precursor disease, IgM monoclonal gammopathy of undetermined significance (MGUS) and active WM flanking the intermediate state of smoldering WM.2 This uncommon lymphoma accounts for approximately 1,500–2,000 new cases a year in the United States, with a median age at presentation of approximately 70 years and a median overall survival (OS) approaching 8–10 years from diagnosis.3–5 Patients may be asymptomatic with an incidental diagnosis, a state referred to as smoldering WM, which accounts for nearly one-fifth of all initial presentations. This subset of WM can be observed without initiation of systemic therapy. In fact, the approach of active surveillance has not demonstrated any adverse impact on the clinical course of patients with smoldering WM.6
The consensus criteria for the initiation of systemic therapy in WM, originally proposed in the Second International Workshop for Waldenström Macroglobulinemia Workshop in 2002, still continue to guide clinicians regarding the appropriate indications for commencing therapy in WM.7 The factors that most commonly require initiation of therapy include significant fatigue, moderate to severe anemia (<10g/dL) or thrombocytopenia (<100x 109/L), bulky symptomatic lymphadenopathy, symptomatic organomegaly, moderate to severe peripheral neuropathy, symptomatic cryoglobulinemia, cold agglutination disease and symptomatic hyperviscosity.8 WM-associated symptomatic hyperviscosity is present in approximately 10–30% of patients at diagnosis or can occur later in the course of disease. Typically, WM-associated symptomatic hyperviscosity manifests as headaches, retinal hemorrhage or retinal vessel tortuosity, epistaxis, gum bleeding, and, rarely, confusion, among others.9–11 WM can infrequently (7–10%) be diagnosed in the presence of a co-existing light-chain (AL) or heavy-and-light-chain (AHL) amyloidosis, which in turn can result in cardiac or renal dysfunction, or other structural and functional organ damage, and thus conferring worse survival in this subset of patients.12,13 Moderate to severe peripheral neuropathy is an indication for treatment initiation in WM.7 Histological transformation to an aggressive lymphoma is an uncommon complication in WM, encountered in 4–6% of patients and typically developing 4–5 years after the initial diagnosis.14,15 Another uncommon complication seen in WM is Schnitzler syndrome, which can manifest as a pruritic urticarial rash commonly accompanied by fever, joint pains and elevated erythrocyte sedimentation rate believed to be due to an autoimmune inflammatory response to the IgM protein.16 This review highlights the significant strides that have been made toward understanding the disease biology and the resultant expansion of therapeutic options in WM in recent years.
Mutational profile in Waldenström macroglobulinemia
The data from the whole genome sequencing in patients with WM show mutations in the MYD88, CXCR4 and ARID1A genes as the most frequently encountered genetic alternations.17 Among the cytogenetic abnormalities, chromosome 6q deletions and trisomy involving chromosomes 4, 12 or 18 represent the most frequently encountered chromosomal alterations in WM, with 6q deletion noted in up to 50% of cases.18,19
MYD88 mutation
The most common somatic mutation in WM is a point mutation (L265P) in the gene coding for the myeloid differentiation primary response 88 (MYD88) protein associated with the B-cell receptor pathway and is encountered in 80–95% of WM cases.20–22 The mutation in the MYD88 gene leads to a constitutive homodimerization of the MYD88 adaptor protein, resulting in downstream signaling through Bruton tyrosine kinase (BTK), interleukin-1 receptor-associated kinase 1 (IRAK1)/IRAK4 kinases and transactivation of hematopoietic cell kinase (HCK), ultimately leading to WM cell survival and proliferation through upregulation of the nuclear factor kappa beta (NFκB) pathway.23 This mutation is not pathognomonic of WM and is observed, albeit in lower rates, in other malignancies including central nervous system lymphoma and diffuse large B-cell lymphoma, and infrequently in splenic marginal zone lymphoma and chronic lymphocytic leukemia.24,25 Nonetheless, the presence of MYD88L265P mutation aids hematopathologists in honing the diagnosis of WM in appropriate clinicopathologic settings. In current clinical practice, the MYD88L265P mutation serves as a diagnostic marker for WM, as well as a predictive marker for BTK-inhibitor monotherapy.26,27 In rare instances, the occurrence of non-L265P mutations in the MYD88 gene has also been noted in WM, and the presence of such mutations is also associated with disease that is responsive to ibrutinib.26
Patients with MYD88L265P mutated genotype tend to have a higher risk of histological transformation to a more aggressive lymphoma, and interestingly, a shorter time-to-progression from smoldering WM to symptomatic disease.20,28 The impact of MYD88L265P mutation status on the OS of patients with WM is debatable, with contradictory reports in the literature; one suggesting inferior OS with the MYD88WT genotype29 and other studies, including the one from Mayo Clinic, suggesting no impact of MYD88L265P status on the OS.10,30 In the Mayo Clinic study, patients were predominantly treated with non-BTK inhibitor based regimens, and OS was calculated from the time of active disease.20
CXCR4 mutation
Alternations in the C-terminal of the CXCR4 gene account for the second most common somatic mutations in WM, and are observed in 30–40% of patients.17,30 These mutations are similar to the germline mutations in the CXCR4 gene, noted in a rare congenital immune deficiency syndrome presenting with warts, hypogammaglobulinemia, infections and myelokathexis (WHIM) syndrome, hence the term CXCR4WHIM mutations. These mutations are broadly grouped as nonsense (NS) mutations or frameshift (FS) mutations. The mutations in the C-terminal region leave the ligand binding region of the CXCR4 receptor intact and prevent internalization of the receptor upon binding with the SDF1 ligand. This results in sustained downstream signaling through the AKT, extracellular signal-regulated kinase (ERK) and BTK pathways.31
Patients with CXCR4WHIM/NS mutations present with a higher degree of marrow lymphoplasmacytosis compared to CXCR4WHIM/FS and CXCR4WT mutations. A retrospective study demonstrated CXCR4 mutated status as a predictor of a shorter treatment-free interval for smoldering WM compared to a CXCR4WT cohort, but CXCR4 status has not been shown to be prognostic for OS in patients with WM.30 The CXCR4WHIM/NS mutation is associated with increased resistance to ibrutinib, as well as other agents such as venetoclax and ixazomib.32–34 The presence of these mutations however does not have an unfavorable impact on OS. Therefore, the utility of evaluating them in the routine clinical practice is currently unclear outside of patients treated in clinical trials or with BTK inhibitors.35
Frontline therapy for Waldenström macroglobulinemia
The therapeutic options for WM have expanded substantially over the past decade. Alkylator monotherapy and purine analog-based therapies remained the cornerstone of therapy for WM until the early 2000s.27 The WM1 study comparing fludarabine with chlorambucil as frontline therapy for WM demonstrated significant improvement in OS with oral fludarabine.36 However, both purine-analogs and alkylator monotherapies or certain combinations (e.g. cyclophosphamide, doxorubicin, vincristine, and prednisone [CHOP]/cyclophosphamide, prednisone and vincristine [CPV]) have fallen out of favor, owing to higher rates of toxicities.37,38 The introduction of less toxic and efficacious rituximab-alkylator combinations, and more recently, the introduction of BTK-inhibitors, has dramatically changed the frontline therapy landscape for WM.39
Rituximab is an anti-CD20 monoclonal antibody, and as monotherapy demonstrates an overall response rate (ORR) of 25–40%, but is less frequently used at academic institutions in the current era due to inferior responses compared to rituximab-based combination therapy and a higher rate of IgM flare (an increase in IgM levels by at least 25% from baseline).40,41 Rituximab monotherapy may still be appropriate in frail patients or in those with a low disease burden who do not require rapid cytoreduction and debulking, but, in the vast majority of patients, rituximab based combinations are the preferred frontline therapeutic options in WM.42
Therapeutic plasmapheresis is recommended as a temporizing measure for the acute management of symptomatic hyperviscosity and needs to be followed by initiation of some form of systemic therapy targeting the lymphoplasmacytic clone.43
The currently preferred frontline therapeutic options for WM can be broadly classified as rituximab-alkylator combination therapy (chemoimmunotherapy), BTK-inhibitor based therapy or proteasome-inhibitor (PI) based therapy.
Chemoimmunotherapy
With the integration of rituximab in the management of CD20 positive lymphomas, rituximab-alkylator combinations (chemoimmunotherapy) became the natural choice for frontline therapy in WM. Currently, the two most commonly used chemoimmunotherapy regimens incorporated in the treatment of WM include dexamethasone, rituximab and cyclophosphamide (DRC) and bendamustine-rituximab (BR).1 The DRC regimen, initially proposed by the Greek group in a phase II study, consisted of six 21-day courses of dexamethasone 20 mg intravenously (IV), followed by rituximab IV 375 mg/m2 and oral cyclophosphamide 100 mg/m2 twice daily (days 1 to 5). A total of 72 patients were included in the study and demonstrated an ORR of 83% with 67% patients achieving a partial response (PR); the 2-year progression-free survival (PFS) and OS were 67% and 90%, respectively.44 In an updated analysis with a median follow-up of 8 years, the median PFS was 35 months (95% confidence interval (CI): 22–48 months) but the median time-to-next therapy (TTNT) was substantially longer at 51 months, suggesting that patients with WM with biochemical progression can safely be observed until the development of symptoms or significant cytopenias for initiation of salvage therapy. The median OS was 95 months (95% CI: 87–103 months). Secondary myelodysplastic syndrome was noted in 3% of patients.45
Subsequently, a multicenter phase III non-inferiority study (the Study Group for Indolent Lymphomas [StiL] trial), compared rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) therapy with BR (bendamustine 90 mg/m2 day 1,2 and rituximab 375 mg/m2 on day 1 of each cycle, administered every 28 days) as frontline therapy for indolent lymphomas, including follicular, lymphoplasmacytic (WM), marginal zone, small lymphoplasmacytic or mantle cell lymphoma. In a subset of patients with WM (n=41, 8% of the study population), the BR arm demonstrated a significantly longer PFS [69.5 months (95% CI: 37–73)] compared to R-CHOP [28.1 months (18–51), hazard ratio 0.33; p=0.003] despite similar ORR with both the regimens.46 The toxicity profile of BR was also different as compared to R-CHOP.
DRC and BR represent two well tolerated and efficacious regimens for the treatment of WM. However, there are no prospective data comparing the two regimens. A retrospective study from Mayo Clinic demonstrated a trend toward longer PFS with BR compared to DRC in both the treatment-naïve setting (2-year PFS 88% versus 61%, p=0.07) as well as in the relapsed/refractory setting (58 months versus 32 months; p=0.08) for patients with WM.47 This finding of superior PFS with BR compared to DRC has subsequently been confirmed in another single-center retrospective study looking at outcomes in the treatment-naïve setting.48 The Mayo study also suggested efficacy of DRC and BR (non-BTK inhibitor based regimens) in MYD88WT patient population. However, the lack of prospective data limits the strength of the findings from this study.47
Our preferred approach is to use BR for a total of four to six 28-day cycles.1 The administration of rituximab, particularly as monotherapy has been associated with an IgM flare phenomenon, wherein there is sudden rise in IgM level after administration of rituximab and can result in the development of symptomatic hyperviscosity.49 This phenomenon results from interleukin-6 (IL-6) secretion by monocytes that are activated by the Fc portion of rituximab antibody and in turn stimulates IgM secretion by WM cells.50 It is less frequently encountered with rituximab-based combination chemotherapy. However, rituximab may be omitted from the initial one or two cycles of the combination regimens, particularly in patients with a high IgM level (>4000 mg/dL) at treatment initiation, to avoid flare associated hyperviscosity.
Bruton tyrosine kinase inhibitor-based therapies
The discovery of the MYD88 mutation in WM has been a seminal event in shaping the treatment landscape for patients with WM. Ibrutinib is a first-in-class oral irreversible inhibitor of BTK which functions as a B-cell receptor signaling kinase, resulting in downstream activation of NFκB, which in turn promotes cell proliferation and survival. Ibrutinib was first studied in a relapsed/refractory WM population and demonstrated substantial activity in MYD88MUTpatients26 that resulted in its approval by the US Food and Drug Administration for WM. In the treatment-naïve setting, the ORR (minor response or better) and major response rate (MRR; partial response or better) with ibrutinib monotherapy (n=30) were 100% and 83%, respectively, and all patients were MYD88MUT genotype.51 However, complete responses with ibrutinib have been elusive. The 18-month estimated PFS for patients treated with ibrutinib in the frontline setting (n=30) was 92% and all patients were alive after a median follow-up of 14.6 months.51 Additionally, the patients with the CXCR4WT genotype demonstrated a higher MRR compared to patients with CXCR4MUT genotype.26,51 A recent single-center retrospective study demonstrated that patients with CXCR4WHIM/NS mutations have an inferior progression free survival (PFS) with ibrutinib compared to CXCR4WHIM/FS and CXCR4WTmutated patients.52
Recently, a phase III randomized trial (iNNOVATE study) compared ibrutinib in combination with rituximab (IR) with rituximab monotherapy plus placebo in patients with treatment-naïve or relapsed WM that were not refractory to prior rituximab therapy. The primary end point for the study was PFS. In the treatment-naïve cohort (n=68), IR demonstrated a significantly improved 24-month PFS of 84% compared to 59% in the placebo-rituximab arm (hazard ratio 0.34; 95% CI: 0.12–0.95). The improvement in PFS was observed irrespective of the patients’ MYD88L265P and CXCR4 genotypes.54 Previous reports have suggested inferior response rates and outcomes for CXCR4MUT patients compared to the CXCR4WT genotype with ibrutinib monotherapy.51–53 However, in this phase III study, the 30-month PFS was 80% in both CXCR4MUT and CXCR4WT cohorts, suggesting that the addition of rituximab may overcome the resistance to ibrutinib in CXCR4MUT genotype.51 The combination of ibrutinib with rituximab also reduced the rate of IgM flare compared to that seen in the placebo-rituximab arm (8% versus 47%), likely due to reduction in the circulating cytokines with concurrent ibrutinib use.54
The substantial responses seen with ibrutinib therapy do come at the cost of unique toxicities and indefinite therapy. Apart from cytopenias, atrial fibrillation (AF) is a common complication with ibrutinib therapy, and grade 3 or more AF can be seen in 10–12% of patients, often requiring dose reduction or permanent discontinuation.53,54 Hypertension has also been described as a notable complication of ibrutinib therapy in 5–10% of cases.53,54 Major hemorrhages have been noted with ibrutinib, but they are uncommon and seen in fewer than 5% of cases.53,54 Recently, a ‘rebound phenomenon’ that results in a rapid rise in serum IgM levels has been described in roughly one-third of cases after abrupt discontinuation of ibrutinib and can result in symptomatic hyperviscosity.55,56 Acalabrutinib, a potent next-generation BTK inhibitors, has shown an ORR of >90% in both treatment-naïve setting (n=14) with 24-month PFS and OS rates of 90% and 92%, respectively.57
Proteasome inhibitor-based therapy
Proteasome inhibitor-based therapy represents another important treatment option for WM. Most notably, bortezomib-dexamethasone-rituximab (BDR) has been used in the frontline setting. A phase II study of BDR in 59 patients with treatment-naïve symptomatic WM demonstrated an ORR of 85% and an MRR of 64%.58 BDR was administered as a single 21-day
cycle of bortezomib alone (1.3 mg/m2 IV on days 1, 4, 8, and 11) for cycle 1. Cycle 2 to 5 comprised of weekly intravenous bortezomib (1.6 mg/m2 on days 1, 8, 15, 22 x 4 35-day cycles) with intravenous dexamethasone (40 mg) and intravenous rituximab (375 mg/m2). After an extended follow-up, the median PFS was 43 months with a 7-year OS of 66%. Peripheral neuropathy was noted in 46% patients (7% grade 3) and dose reduction by at least one level was required in 37% patients.59 In 8% of patients, bortezomib was discontinued altogether due to neuropathy but resolution to grade 1 or less was noted in most of the patients over time. Additionally, secondary myelodysplastic syndrome was not observed.59
In our practice, bortezomib-based regimens are less favored due to high rates of development or worsening of peripheral neuropathy in patients with a pre-existing neuropathy which can be encountered in up to 20% of patients with WM.58,60 However, in the initial phase II studies, bortezomib was administered intravenously, leading to potentially higher rates of neuropathy than would be expected with subcutaneous administration, which is the current standard in multiple myeloma.60 Notably, in a phase II study incorporating subcutaneous bortezomib, cyclophosphamide and rituximab (BCR), no grade 3 or higher rate of peripheral neuropathy was noted, making this route of administration preferable in this neuropathy-prone patient population going forward.61
A phase II study looked at the combination of carfilzomib, rituximab and dexamethasone (CaRD) in rituximab and carfilzomib naïve patients with WM that had previously not received any treatment for more than a year. The CaRD regimen demonstrated an ORR of 87% and MRR of 68%. Cardiomyopathy and neuropathy were noted in 3% each. The median PFS for the cohort was 46 months (range 2–63 months) but the OS data are currently immature.62,63Ixazomib, an oral proteasome inhibitor, has been studied in combination with rituximab and dexamethasone (IDR) in a recent small phase II study of 26 treatment-naïve patients with symptomatic WM. The IDR regimen demonstrated an ORR of 96% and MRR of 77%. Grade ≥3 neuropathy was noted in only 4% with any grade neuropathy in 23%. Outcome data are not yet mature for this study.34
Longer follow-up is required to assess the outcomes with these newer PI-based regimens. Modified schedule (weekly instead of twice weekly) and route of administration (subcutaneous instead of IV) of bortezomib, or incorporation of less neurotoxic agents such as carfilzomib or ixazomib in place of bortezomib, are strategies to reduce the risk of treatment-emergent neuropathy with PI-based regimens in WM.34,62,64
Factors guiding selection of frontline therapy for Waldenström macroglobulinemia
As described previously, chemoimmunotherapy or BTK inhibitor-based therapies represent the two most commonly used approaches in WM, with stark differences between them in terms of the duration of therapy, routes of administration and toxicity profiles of the regimens. The decision for choosing frontline therapy is primarily based on patient preference or the presence of comorbidities, and the genotype (primarily the MYD88 genotype, with some data emerging for CXCR4 genotype in guiding therapy).
BTK inhibitors come with the obvious advantage of being available as oral formulations. This increases convenience in terms of drug delivery and is likely to be appealing to patients. The traditional chemotherapeutic side effects like nausea, vomiting and profound cytopenias are less common with ibrutinib although a head-to-head comparison in WM has not been undertaken.1,27 Also, IgM flare is not encountered with ibrutinib monotherapy. However, as discussed earlier, BTK inhibitors come with a unique toxicity profile, including atrial fibrillation and hypertension, which may pose a challenge, particularly in elderly patients who make up the predominant population in this disease. The two most important limitations with ibrutinib monotherapy are the need for continuous therapy until progression or intolerable side effects, and lower efficacy in patients MYD88WT genotype.26,65 A prolonged treatment duration can have significant financial implications. In addition, the abrupt discontinuation of this agent is associated with a rebound increase in IgM, something that is not observed with traditional chemoimmunotherapy approach.
There is also emerging evidence to suggest that patients exhibiting the CXCR4MUT genotype, specifically those with nonsense mutations, can demonstrate increased resistance and shorter PFS rates when treated with ibrutinib monotherapy.32,52,53 Thus, patients with a CXCR4MUT genotype needing a rapid response may be preferentially treated with chemoimmunotherapy or PI-based therapy, although the adverse impact of CXCR4MUT is overcome with the use of ibrutinib with rituximab, a regimen that appears to exhibit equivalent response rates, irrespective of the CXCR4 genotype.54 The impact of CXCR4 mutation on outcomes for patients treated with chemoimmunotherapy is currently lacking and thus the utility of CXCR4 mutation assessment in guiding therapy is currently limited.
Chemoimmunotherapy is typically given for a fixed duration of 4–6 months, providing a treatment-free interval for patients, but can be associated with an increased risk of myelodysplastic syndrome in the future.45,66
We have proposed an algorithm to aid in choosing frontline therapy in WM based on the urgency of therapy, duration of therapy and the MYD88 mutational status (Figure 1).
Figure 1. Choice of frontline therapy for symptomatic Waldenström macroglobulinemia
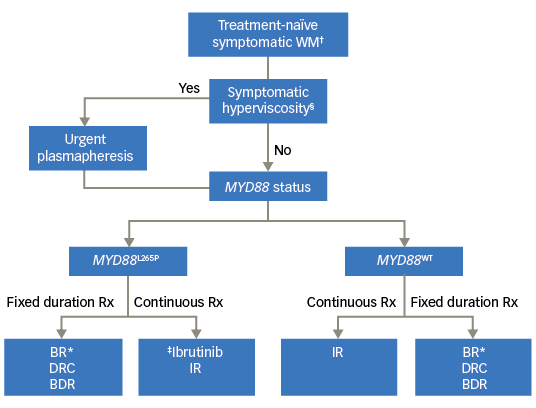
BDR = bortezomib, dexamethasone, rituximab; BR= bendamustine rituximab; DRC = dexamethasone rituximab, cyclophosphamide; IR = ibrutinib plus rituximab; Rx = prescription. †Patients meeting the 2002 Consensus criteria for treament initiation; §Presence of one or more hyperviscosity-related clinical symptoms such as bleeding, blurry vision, headache, vertigo, dizziness, nystagmus, deafness, slow mentation, changes in retinal blood vessels, or ataxia in patients with WM that was otherwise not attributable to another cause; *Our preferred regimen is BR (over DRC or BDR); ‡No prospective study comparing ibrutinib versus BR/DRC/BDR; BDR to be avoided in patients with pre-existing neuropathy.
Management of peripheral neuropathy in Waldenström macroglobulinemia
The pathogenesis of neuropathy in WM is unclear but is, at least in part, attributed to the involvement of circulating IgM protein in demyelination.67 The IgM protein binds to myelin-associated glycoprotein (MAG). Anti-MAG antibodies, and less commonly, anti-ganglioside antibodies, have been found to be elevated in patients with WM and peripheral neuropathy.68,69 Associated conditions like AL amyloidosis and polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS) syndrome, can also present with neuropathy in the presence of a monoclonal protein, and represent important differential diagnoses that need to be ruled out.70,71
Apart from symptomatic treatment of neuropathy, initiation of systemic chemotherapy and rituximab can potentially benefit the neuropathy associated with WM.67 In a meta-analysis that included two randomized trials assessing the role of rituximab in patients with anti-MAG antibodies, some benefit of rituximab therapy was noted with regard to improvement of the neuropathy disability scales.72 This data is however of limited strength and would benefit from additional studies being carried out. On the other hand, use of cyclophosphamide failed to show any significant benefit in anti-MAG associated neuropathy.72
With regard to the role of the BTK inhibitor ibrutinib in the setting of peripheral neuropathy, the pivotal trial studying the role of this agent in a relapsed/refractory setting (n=63) included nine patients (14%) with peripheral sensory neuropathy related to the IgM protein.53 Of these nine patients, five had a subjective improvement following initiation of ibrutinib, and neuropathy symptoms stabilized in the remaining four patients, suggesting that ibrutinib may be a safe option in this setting.53 However, the study lacked objective data such as utilization of the neuropathy impairment score or periodic nerve conduction studies to document stabilization or improvement in peripheral neuropathy.53 Care should be taken to avoid bortezomib-based regimens.
Maintenance rituximab in Waldenström macroglobulinemia
There is currently limited data addressing the role of maintenance rituximab in patients with WM. In a single-center retrospective study of 248 patients, 86 (35%) rituximab-naïve patients received maintenance rituximab. The cohort receiving maintenance rituximab demonstrated an improvement in PFS and importantly OS as well, albeit at the cost of higher rate of infections.73 A large prospective randomized trial (Significance of Duration of Maintenance Therapy With Rituximab in Non-Hodgkin Lymphomas [MAINTAIN] study; NCT00877214) is currently underway for assessing the role of maintenance therapy with rituximab in indolent lymphomas, including WM treated with BR as primary induction therapy. The primary endpoint of this study is PFS.
In the absence of a prospective study and additional toxicities associated with maintenance rituximab, our current approach is to watch and wait after induction therapy. It is also important to understand that while improvement in PFS is likely to be demonstrated with maintenance rituximab, it may not be an ideal benchmark for justifying use of maintenance rituximab. In an indolent disease like WM, it is reasonable to wait and watch patients after induction therapy until the recurrence of symptoms when a salvage therapy can be reinstituted. Thus, time to next therapy (TTNT) and OS are likely to be better measures in assessing the benefit of maintenance therapy in WM. Nonetheless, rituximab maintenance is a promising strategy that needs further study before incorporating it as a standard of care.
Salvage therapy for relapsed/refractory Waldenström macroglobulinemia
Even though WM is an indolent disease with a good survival outcome, the incurable nature of the disease means that relapse after frontline therapy is fairly common. Frequently, the disease can progress biochemically without any symptoms and these asymptomatic patients do not merit initiation of systemic therapy. For this reason, TTNT tends to be longer than the PFS in studies involving patients with WM45 and is likely to be a more practical measure of efficacy for a regimen in an indolent disease.74 However, TTNT can be subjective to biases of the treating physicians and needs to be interpreted with caution. The 2002 consensus criteria for initiation of systemic therapy serves as a good guide to restarting therapy in the relapsed setting as well.7
The selection of therapy in the first relapsed setting may be somewhat more straightforward than the selection of frontline therapy, with preference given to the use of a regimen not previously incorporated in the treatment of WM. The exception to this strategy is if a substantially durable remission was observed with the previously used fixed-duration therapy, in which case consideration for repeating the regimen should be given. If the response duration to the frontline therapy had been considerably long (typically TTNT more than 3–4 years), retreatment with the same regimen may be reasonable.1
Chemoimmunotherapy regimens are safe and effective in relapsed setting as well, with BR demonstrating an ORR and MRR of 80% and 75%, respectively, in a retrospective cohort of 71 patients with relapsed/refractory WM.75 However, care should be taken while using bendamustine in patients who have been previously treated with nucleoside analogs, due to the risk of prolonged cytopenias.76
As highlighted earlier, ibrutinib is efficacious in the relapsed/refractory setting and represents a viable option in patients that have not been exposed to a BTK inhibitor previously.77
The PI-based regimens, including, BDR CaRD and IDR are effective treatment options in the frontline setting for WM that can be utilized in the relapsed setting as well.62,78 In a multicenter study of 27 patients with relapsed/refractory WM, single agent intravenous bortezomib showed an impressive ORR of 85% and a MRR of 48%, albeit with a grade 3 sensory neuropathy development in 22% of patients.79 There is limited evidence suggesting that PI-based therapy may overcome the potentially negative impact of the CXCR4MUT genotype on disease outcomes in WM, although the sample size in these studies is relatively small, which prevents a strong case being made for this treatment approach.62 Oprozomib is another a new oral PI that has been studied in relapsed/refractory WM. A phase II study tested two different dosing regimens, either once a day for five days, every 14 days (5/14 schedule) or on Days 1, 2, 8 and 9, every 14 days (2/7 schedule). The study demonstrated an ORR and MRR of 71% and 50% on the 2/7 schedule and 47% and 29% on the 5/14 schedules, respectively. However, oprozomib was associated with significant grade 3 or more gastrointestinal toxicities, including nausea (13%), diarrhea (33%), and abdominal pain (13%), leading to frequent drug discontinuation.80,81
The ORR and MRR with ibrutinib in MYD88MUT cohort were 100% and 92% in the relapsed/refractory setting, respectively, whereas in the MYD88WT cohort, ORR and MRR was 60% and 0%, respectively.26 The 2-year estimated PFS and OS with ibrutinib in the relapsed/refractory setting (n=63) were 69% and 95%, respectively.26 Long term follow-up of a another prospective study evaluating relapsed/refractory patients treated with ibrutinib monotherapy demonstrated inferior PFS in the MYD88MUTCXCR4MUT cohort compared to the MYD88MUTCXCR4WT cohort.82 In a pre-specified subgroup analysis of the iNNOVATE phase III trial, ibrutinib-rituximab doublet demonstrated a significant improvement in the 24-month PFS compared to placebo-rituximab (80% versus 37%; hazard ratio 0.17 [95% CI: 0.08–0.36]) in the relapsed/refractory cohort (n=82).54
Multiple next-generation BTK inhibitors that are more specific and result in fewer off-target effects are in advanced stages of development and clinical trials. Acalabrutinib is a highly potent BTK inhibitor that has shown an ORR of >90% in the relapsed/refractory setting (n=92), with 24-month PFS and OS rates of 82% and 89%, respectively in relapsed/refractory WM.57 Zanubrutinib (BGB-3111), another newer generation BTK inhibitor, appears to be a promising agent. It is also associated with fewer off-target effects compared to ibrutinib, which can simultaneously inhibit TEC, interleukin-2 inducible T-cell kinase (ITK) and epidermal receptor growth factor (EGFR) kinases, among others, and is likely responsible for some of the adverse effects including diarrhea and AF.83 In a phase I study that enrolled 46 patients with relapsed WM, zanubrutinib demonstrated excellent efficacy with an ORR of 90% and MRR of 76%. The increase in the very good partial response (VGPR) rate (43% with zanubrutinib) in comparison to that reported initially with ibrutinib (16%) was particularly notable. The adverse event profile included a lower rate of AF (6%, 3/46) and grade 3 diarrhea (2%). The most frequent complications were mostly grade 1 or 2 upper respiratory infections (33%), contusion (28%) and constipation (22%).84 A randomized phase III study comparing zanubrutinib and ibrutinib in patients with WM has completed enrollment and the results are awaited (Table 1).85
Targeting the PI3K/AKT/MTOR by everolimus has also demonstrated responses in WM in both frontline and relapsed setting, but is associated with a high rate of adverse effects, including mucositis, diarrhea and life-threatening pneumonitis.86,87 It should therefore be avoided in the frontline or the initial relapsed setting for which a multitude of options exist.
Lenalidomide has been studied in combination with rituximab as well as DRC (lenalidomide, rituximab, cyclophosphamide, and dexamethasone [LR-CD] regimen) in frontline and relapsed and/or refractory settings, and has demonstrated good clinical activity.88,89 However, a previous study of lenalidomide in combination with rituximab for the treatment of WM led to unexpected high rates of anemia, even with low doses of lenalidomide, resulting in premature closure of the study.90 Apart from rituximab, obinutuzumab and ofatumumab represent other CD20-targeting strategies that may be utilized in WM.27,91
Autologous stem cell transplant (ASCT) represents another potential option in the treatment of WM.92 Although the 5-year PFS and OS rates of 40% and 70% in the European Group for Blood and Marrow Transplantation (EBMT) 158-patient cohort was impressive, it came at the cost of an increase in the rate of secondary cancers (8.4% at 5 years) and a non-relapse mortality of ~4% at 1 year.93 Additionally, the outcomes for ASCT are best when it is incorporated earlier in the relapsed setting rather than in refractory WM or following multiple relapses.27 The availability of more efficacious and less toxic treatment options appears to be limiting the utilization of ASCT to the setting of AL amyloidosis or histological transformation in WM. Nonetheless, it needs to be noted that stem cell collection is advisable after the initial few cycles of frontline therapy if an autologous stem cell transplant is being considered in the future, especially for young patients with WM.92 The disease course of WM can be uncommonly complicated by transformation to an aggressive histology, most commonly diffuse large B-cell lymphoma (DLBCL).14 The treatment of transformed WM is similar to the approach for patients with DLBCL. The initial induction therapy is typically with R-CHOP or a similar regimen, followed by either observation or ASCT. There are currently no conclusive data to suggest superiority of a specific approach over other approaches.15,94
Allogeneic stem cell transplant may also have a role in management of transformed WM.95 By contrast, the use of allogeneic stem cell transplantation in patients with WM without transformation is discouraged outside of clinical trials. A recent study conducted by the Center for International Blood and Marrow Transplant Research (CIBMTR) reported outcomes of 144 patients undergoing allogeneic transplant for relapsed WM.96 Nearly 50% of the patients (n=67) received myeloablative conditioning whereas the rest were conditioned with a reduced-intensity regimen. The 5-year PFS and OS rates were 46% and 52%, respectively, with a high 5-year non-relapse mortality rate of 30%. Chemosensitive disease at transplant was associated with a better OS.96 Our current approach is to not consider allogeneic transplantation for WM outside of a clinical trial.
More novel combinations and therapy targets are in the pipeline for WM. Inhibition of Bcl-2 using venetoclax appears to be especially promising.97 In a phase II study of venetoclax in 30 patients with previously treated WM, an ORR of 80% and MRR of 53% were noted. The VGPR rates was lower (6%) in patients with CXCR4MUT genotype compared to those with the CXCR4WT genotype (29%). Notably, all patients in this study carried MYD88L265P mutations, thus currently limiting the applicability of this data to the MYD88MUT genotype.33
Targeting the immune checkpoints through programmed death-ligand 1 (PD-L1) inhibition is also a promising option and studies are currently underway (Table 1).
Patients with WM appear to be well suited for chimeric antigen receptor (CAR) T cell therapies, with initial constructs using CD19-directed CAR-T cell showing promising preclinical activity in WM.98
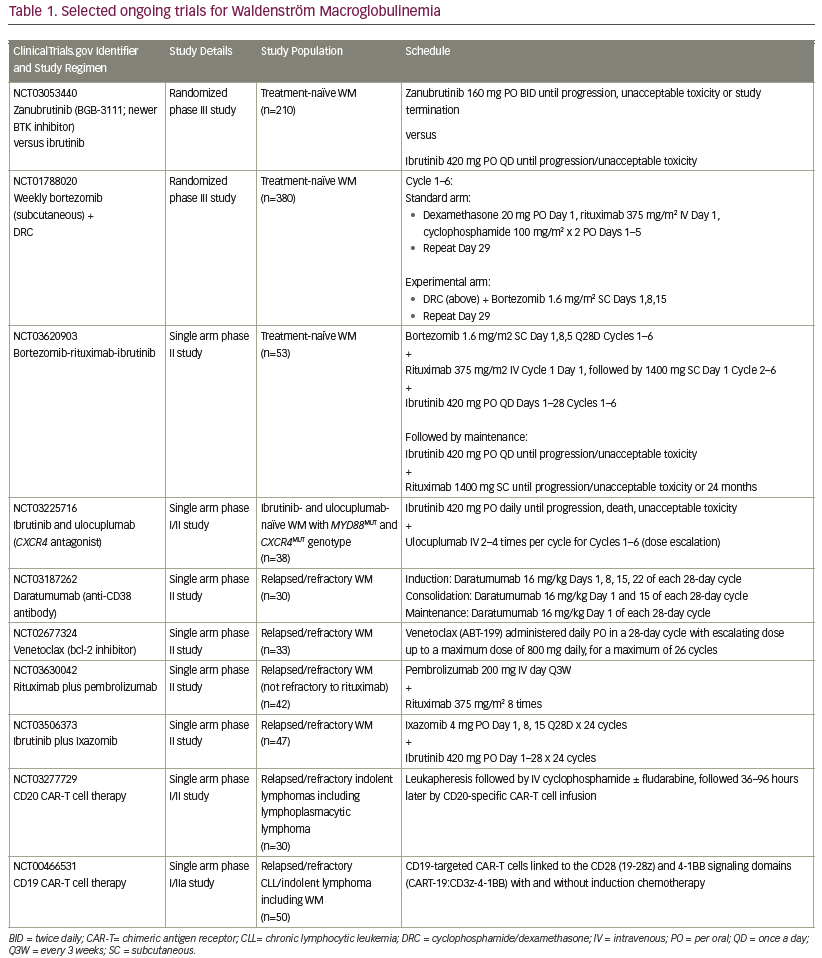
Newer approaches, including targeting the CXCR4 or toll-like receptors are under investigation as potential therapies for WM. A list of selected recently proposed/ongoing studies specific for patients with WM is outlined in Table 1.
Purine analogs and alkylators, either alone or in combination with an anti-CD20 antibody, still hold a place in the management of patients
with multiply relapsed refractory disease. Enrollment in clinical trials is highly encouraged in all settings particularly for the relapsed-refractory patient population.
Concluding remarks
With the continuous improvement and deepening of our understanding of the biology of this fascinating disease, there is an increasing effort to provide a more personalized therapy for patients with WM. As we continue expand our therapeutic options, care should be taken to avoid overtreatment in this typically indolent malignancy.