Bladder cancer is one of the most common malignancies in the USA, with an estimated 81,180 new cases and 17,100 deaths in 2022.1 Advanced urothelial carcinoma (aUC) includes both locally advanced, unresectable and metastatic urothelial cancer. About 5% of all bladder cancer cases present as de novo aUC, while almost 25% of patients present with muscle-invasive bladder cancer (MIBC). About 50% of these patients progress to aUC even with intent-to-cure locoregional therapy, for example, neoadjuvant cisplatin-based chemotherapy (in fit patients) followed by radical cystectomy and pelvic lymph node dissection.2 The treatment landscape in aUC is rapidly evolving with the advent of novel therapeutic agents and approaches. Enfortumab vedotin (EV) is one of six new medications that have been approved by the US Food and Drug Administration (FDA) for aUC since 2017 and has been shown to have high efficacy with acceptable toxicity in clinical trials and retrospective studies.3–7 This article will briefly review the treatment landscape of aUC in both the first-line and salvage settings. We will then focus on EV in the treatment landscape of aUC and, in particular, on the landmark phase III EV-301 trial.5
Methodology
A literature search was performed by the authors from March to May 2022 through PubMed/Medline to identify available articles and studies pertaining to the biology and systemic treatment of MIBC and aUC. The search was performed using the following keywords, separately and in different combinations: “bladder cancer”, “antibody drug conjugate”, “checkpoint inhibition”, “metastatic urothelial carcinoma”, “immunotherapy”, “Sacituzumab govitecan”, “Erdafitinib”, “Enfortumab vedotin”, “target therapy” and “trial”. Current abstracts with preliminary data from international conferences, such as the 2022 American Society of Clinical Oncology (ASCO) Genitourinary Cancers Symposium, 2022 ASCO Annual Meeting and the 2022 European Society for Medical Oncology Congress, were reviewed and used for this systematic review. The authors performed the study selection following the recommendations of the Preferred Reporting Items for Systematic Review and Meta-analysis Statement.8
First-line treatment options
First-line therapy for aUC includes platinum-based chemotherapy followed by switch maintenance avelumab for those with a response or stable disease after 4–6 cycles of chemotherapy. For patients deemed fit for cisplatin, cisplatin-based combinations, such as gemcitabine with cisplatin or dose-dense methotrexate, vinblastine, doxorubicin, cisplatin (MVAC), are preferred and have demonstrated overall response rates (ORRs) of up to 50–55%, including approximately 10–15% complete responses (CRs).9 Initially, cisplatin was considered the standard of care in patients with aUC as early prospective studies showed remissions ranging from 30 to 40%.10–12 A phase III trial investigating cisplatin alone compared with MVAC showed superior response rates for MVAC (39%) compared with cisplatin alone (12%) and improved overall survival (OS) with median of 12.5 versus 8.2 months, respectively.13 This trial established cisplatin-based combination to be the standard-of-care therapy. However, the conventional MVAC combination was associated with increased treatment-related adverse effects such as neutropenia, neutropenic fever and mucositis. A subsequent phase III clinical trial investigating the conventional MVAC treatment versus cisplatin/gemcitabine showed similar OS between the two regimens (hazards ratio [HR] 1.04; 95% confidence interval [CI] 0.82–1.32; p=0.75), but cisplatin/gemcitabine showed a better safety profile with fewer rates of neutropenia and neutropenic fever.14 The phase III clinical trial EORTC-30924 also demonstrated decreased toxicity and the ability to deliver twice the dose of cisplatin and doxorubicin with dose-dense MVAC, with improved progression-free survival (PFS) and ORR compared with MVAC.15
Almost half of the patients with aUC are unfit for cisplatin-containing chemotherapy due to impaired renl function, poor performance status, symptomatic heart failure, severe underlying neuropathy or hearing loss.16 For these patients, carboplatin with gemcitabine is the preferred first-line regimen, with an ORR of approximately 40%.17 Despite initial chemosensitivity, historical outcomes after these platinum-based regimens have been poor, with median PFS (mPFS) of only 6–8 months and median OS (mOS) of 9–14 months. Historically, only 15% of patients have experienced 5-year OS, and the prognosis is particularly poor among patients with visceral metastases for whom the 5-year OS rate has been below 10%.18 In an effort to improve these outcomes, multiple studies have investigated either biomarkers to aid in patient selection or combination therapies,19,20 including chemotherapy with immune checkpoint inhibitor (ICI) and ICI combinations; unfortunately, these trials have not shown a significant OS benefit over platinum-based chemotherapy.21–24 An approach that has been shown to be very successful is the switch-maintenance immunotherapy. The JAVELIN Bladder 100 phase III trial showed that the use of switch-maintenance avelumab, an anti-programmed death-ligand 1 (PD-L1), plus best supportive care in patients who had a response or stable disease with first-line platinum-based chemotherapy (cisplatin/gemcitabine or carboplatin/gemcitabine) had significantly prolonged OS and PFS compared with best supportive care alone.25
ICIs have also been investigated and approved in the first-line setting for patients who are not eligible to receive platinum. Atezolizumab was FDA approved for aUC in the first-line setting in patients with PD-L1-high tumours (evaluated using the Ventana SP142 assay [Ventana Medical Systems, Inc.; Tucson, AZ, USA]) who were not eligible to receive cisplatin or for those who were not eligible to receive platinum.26 This approval was based initially on the single-arm phase II IMvigor210 cohort one trial, which showed an ORR 0f 23% with durable responses and favourable toxicity profile.27 The ORR was later confirmed in the IMvigor130 phase III trial (ClinicalTrials.gov Identifier: NCT02807636); however, atezolizumab did not significantly prolong OS versus chemotherapy.21 The indication for atezolizumab in the first-line setting was recently withdrawn.28 Pembrolizumab was initially approved in this first-line setting only for patients who were not eligible to receive cisplatin based on the single arm phase II KEYNOTE-052 trial (ClinicalTrials.gov Identifier: NCT02335424), which showed an ORR of 29%, with durable responses and favourable toxicity profile.29 The subsequent KEYNOTE-361 randomized phase III trial (ClinicalTrials.gov identifier: NCT02853305) did not show longer survival with pembrolizumab versus platinum-based chemotherapy, even among those with a PD-L1 combined positive score of ≥10, which led to a revision in the pembrolizumab label by the FDA to limit use to only those who were not eligible for platinum.22,30,31 Several other trials investigating ICI combinations have also been investigated and reported in this setting but have not changed practice.23,32 However, some phase III clinical trials are on-going, including Checkmate 901 (nivolumab/ipilimumab versus platinum-based chemotherapy [ClinicalTrials.gov Identifier: NCT03036098]), NILE (durvalumab with platinum-based chemotherapy versus durvalumab/tremelimumab with platinum-based chemotherapy versus platinum-based chemotherapy alone [ClinicalTrials.gov Identifier: NCT03682068]) and EV-302 (EV with pembrolizumab versus platinum-based chemotherapy [ClinicalTrials.gov Identifier: NCT04223856]).32–34
Novel therapies in the salvage setting
Multiple new therapies have been approved in the salvage setting for patients with aUC who have progressed on first-line therapy. ICIs have been extensively investigated in this setting and have shown durable responses after progression on platinum-based chemotherapy. The phase III KEYNOTE-045 trial (ClinicalTrials.gov Identifier: NCT02256436) demonstrated significantly longer OS with pembrolizumab compared with taxane or vinflunine in patients with progression on first-line platinum-based chemotherapy.35 Atezolizumab, durvalumab, nivolumab and avelumab have also been shown to have durable responses with favourable toxicity profiles compared with historical cytotoxic chemotherapy after progression on first-line chemotherapy in single-arm studies.36–39Pembrolizumab, nivolumab and avelumab have on-going FDA-approved indications for treatment after progression on first-line platinum-based chemotherapy, while atezolizumab and durvalumab also had FDA-approved indications in this setting, although subsequent negative trials led to voluntary withdrawal.23,40–47 However, with the advent of the switch-maintenance therapy avelumab in those with a response or stable disease on first-line platinum-chemotherapy, the utility of second-line ICI has diminished, as many patients receive switch-maintenance avelumab and, thus, require other therapies upon progression.
Erdafitinib, a potent tyrosine kinase inhibitor of fibroblast growth factor receptors 1–4 (FGFR1–4) involved in signalling pathways that regulate cell proliferation, migration and differentiation, has received accelerated FDA approval in patients who progress on platinum-based chemotherapy and have susceptible FGFR2 or FGFR3 alterations (activating mutation or fusion).48 Approximately 20% of patients with bladder cancer and 30–50% of those with upper-tract UC may have such alterations.49–53 The open-label BLC2001 phase II trial in those with susceptible FGFR2 or FGFR3 alterations and documented progression on platinum-based chemotherapy showed an ORR of 40% and an mPFS of 5.5 months with manageable toxicity.54 A follow-up phase III study (THOR) is investigating erdafitinib compared with chemotherapy (vinflunine or docetaxel) or pembrolizumab in patients with aUC with certain FGFR alterations (FGFR3 mutations or FGFR2/3 fusions) in the platinum-refractory setting.55 Erdafitinib was also evaluated in combination with cetrelimab (an anti-PD1) in the first-line setting in the phase Ib/II NORSE trial, with early results showing an ORR of 68%.56
Sacituzumab govitecan is an antibody–drug conjugate targeting Trop-2, an epithelial cell surface antigen highly expressed in UCs. It has shown an ORR of 27%, an mPFS of 5 months and an mOS of 11 months in heavily pretreated patients who had progressed on platinum-based therapy and ICI based on the phase II TROPHY-U-01 trial (cohort 1) and has received accelerated FDA approval in this setting.57 A confirmatory phase III trial (TROPiCS-04 study) is now under way.58 Cohort 3 of TROPHY-U01, which investigated the combination of sacituzumab govitecan with pembrolizumab in the platinum-refractory, ICI-naïve, second-line setting reported an ORR of 34% and a disease control rate of 61% with manageable toxicity.59
There are several reported and on-going trials investigating novel agents, as monotherapy or in combination with ICIs, in patients who have progressed on first-line platinum-based chemotherapy. Targets under investigation include poly (ADP-ribose) polymerase (PARP), FGFR, human epidermal growth factor receptor 2 and mammalian target of rapamycin.60 Below, we review the landmark phase II/III studies investigating novel agents (with or without ICIs) for aUC. The ATLAS trial, an open-label phase II trial investigating rucaparib monotherapy (a PAR inhibitor) in previously treated aUC regardless of homologous recombination deficiency status, did not show activity in unselected patients.61 The open-label multiarm BISCAY trial investigating second-line combination durvalumab with relevant biomarker-selected targeted therapies in patients with aUC who progressed on platinum-based cohorts had four arms:
- combination durvalumab with AZD4547 (an FGFR inhibitor) versus ADZ4547 alone in patients with FGFR3 mutations or FGFR1–3 fusions
- combination durvalumab with vistusertib (TORC1/2 inhibitor) in patients with RICTOR amplification or TSC1/2 mutations
- combination durvalumab plus olaparib (PARP inhibitor) in patients with DDR gene alterations
- durvalumab alone.
Response rates ranged from 9% to 36% across arms and did not meet specified thresholds of target ORR in any of the cohorts.62 Other on-going biomarker-driven studies investigating combination novel agents with ICIs include a phase II trial investigating recombinant albumin fusion protein sEphB4-HSA with pembrolizumab for patients with aUC who had progressed on platinum-based chemotherapy (ClinicalTrials.gov identifier: NCT02717156); the trial demonstrated an ORR of 58%, with an mOS of 15 months in those who expressed EphrinB2.63 The multicohort phase II trial of sitravatinib (a tyrosine kinase inhibitor targeting Tyro3, Axl, and Mer receptors, split family receptors and c-Met) in combination with nivolumab in patients with aUC who had progressed on platinum-based chemotherapy showed an ORR of 31% and an mPFS of 4 months.64 MORPHEUS-mUC (ClinicalTrials.gov identifier: NCT03869190) is a phase Ib/II umbrella trial evaluating the safety and efficacy of atezolizumab in combination with multiple agents with different targets (including Nectin-4, PARP, CD47, CD38, DPP-4 and IL-6R) in platinum-refractory aUC.65
Enfortumab vedotin
Preclinical data and early phase trials
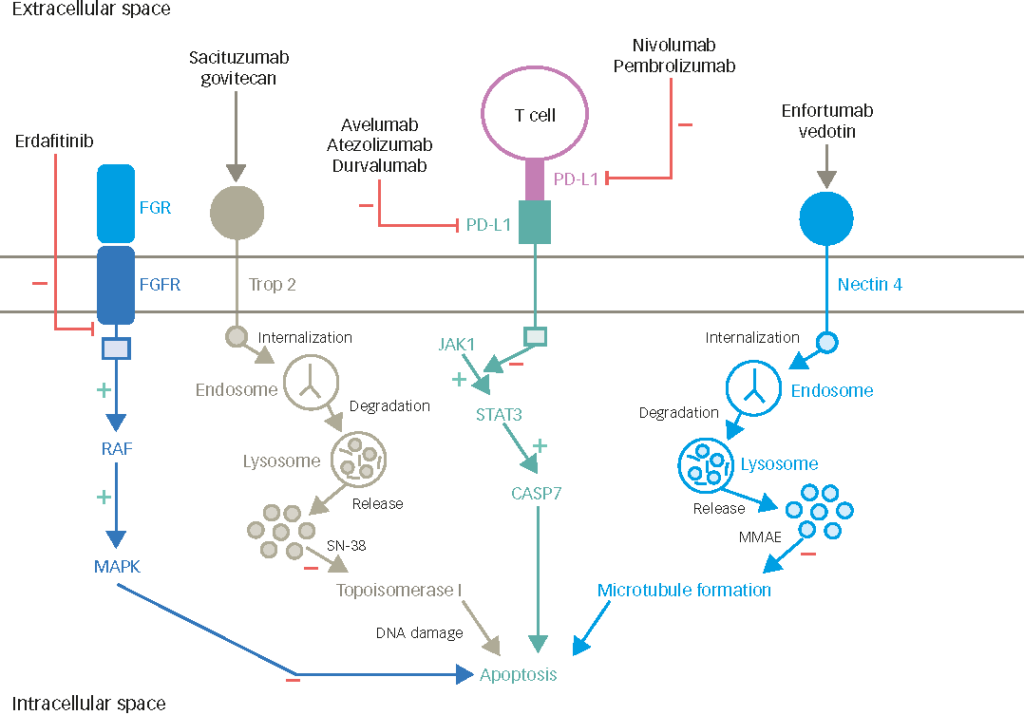
EV is an antibody–drug conjugate comprising a monoclonal antibody targeting Nectin-4, which is covalently linked to the microtubule-disrupting agent monomethyl auristatin E (MMAE). Nectin-4 is a transmembrane protein that belongs to the nectin family of adhesion molecules. It comprises an extracellular domain containing three immunoglobulin-like subdomains, a transmembrane helix and an intracellular region.66 Nectins are thought to mediate Ca2+-independent cell–cell adhesion via both homophilic and heterophilic trans-interactions at adherens junctions and are involved in cell movement and proliferation.67,68 Nectin-4 has been found to be expressed in multiple cancers, particularly urothelial, breast, lung, pancreatic and ovarian cancers.69
In preclinical cancer models, moderate-to-strong Nectin-4 staining was observed in 60% of bladder cancer specimens, 53% of breast cancer specimens, 37% of pancreatic cancer specimens and 27% of lung cancer specimens.69 EV was shown to bind to cell surface-expressed Nectin-4 with high affinity and to induce cell death in vitro in a dose-dependent manner.69 Moderate-to-strong Nectin-4 staining was also observed in 34% of patients with upper tract or renal pelvis UC.70 The current literature evaluating Nectin-4 expression in variant histology bladder cancer is limited. A recent study investigating Nectin-4 expression in bladder cancer revealed that there is a heterogeneity of expression in morphological variants of urothelial cancer and non-urothelial histologic types, with 7/10 (70%) squamous cell carcinomas, 3/11 (28%) micropapillary tumours, 4/6 (66%) adenocarcinomas, 2/4 (50%) nested carcinomas, 5/8 (63%) plasmacytoid, 1/10 (10%) sarcomatoid carcinomas and 0/15 (0%) small cell carcinomas expressing Nectin-4.71
EV-101 (ClinicalTrials.gov identifier: NCT02091999) was a phase I dose-escalation/dose-expansion study in patients with Nectin-4-positive tumours who had progressed on ≥1 line of therapy.72 The trial included 155 patients in the aUC cohort. Results showed that EV was well tolerated, with an ORR of 43% regardless of previous treatment.72
Based on these results, a follow-up single-arm, two-cohort phase II trial, EV-201 (ClinicalTrials.gov identifier: NCT03219333), was undertaken.3 Patients receiving EV alone (cohort 1) demonstrated an ORR of 44% (12% CR), with a median duration of response of 7.6 months in patients with aUC who had progressed after platinum-based chemotherapy and ICI. The most common treatment-related adverse events were fatigue (50%), peripheral neuropathy (50%), rash (48%; 75% of which were grade≤2) and decreased appetite (44%). Treatment-related hyperglycaemia was seen in 11% of patients.4 These results led to the FDA granting accelerated approval to EV in December 2019 for patients with aUC who have received platinum-based chemotherapy and ICI.73
A separate single-arm cohort of EV-201 included patients with aUC who were not eligible for cisplatin and progressed on anti-PD1/L1 therapy.3 In this cohort, the ORR with EV alone was 52% (20% CR rate) with a similar toxicity profile to the one seen in cohort 1. These findings resulted in the expansion of the FDA label in July 2021 (at the same time as the EV regular approval based on the EV-301 trial) to include treatment for patients with aUC, who are ineligible for cisplatin and have received≥1 prior line of therapy.
EV-301
Following the results from EV-201, a confirmatory phase III trial, EV-301 (ClinicalTrials.gov identifier: NCT03474107), was conducted in patients with aUC who had progression on platinum-based chemotherapy and ICI.5 Patients were randomized to receive either EV or chemotherapy (docetaxel, paclitaxel or vinflunine as per the investigator’s choice). The primary endpoint was OS, and other endpoints included ORR, PFS, toxicity and disease control rate.
Overall, 301 patients were randomized to EV and 307 patients to chemotherapy. EV significantly prolonged OS (median 12.8 versus 8.9 months; HR for death 0.70; 95% CI 0.56–0.89; p=0.001) and mPFS (5.5 versus 3.7 months; HR for progression or death 0.62; 95% CI 0.51–0.75; p<0.001) in patients with aUC and progression on platinum-based chemotherapy and ICI. ORR was also significantly higher in the EV group compared with the chemotherapy group (40.6% versus 17.9%, respectively; p<0.001), as well as the disease control rate (79.0% versus 53.4%, respectively; p<0.001). The incidence of treatment-related adverse events was similar between the two groups, with approximately 90% of patients expressing any adverse event and 50% of patients expressing a grade ≥3 event in both groups.5 Specific treatment-related adverse events associated with EV included peripheral neuropathy (46.3% any grade, 5.1% grade≥3; predominantly sensory neuropathy), rash (43.9% any grade, 14.5% grade≥3) and hyperglycaemia (6.4% any grade, 4% grade≥3, including 0.3% grade 5). Early recognition of toxicity and proper management is critical.74 Based on these results, in July 2021, the FDA granted regular approval to EV for patients with aUC after progression on prior platinum-based chemotherapy and ICI.75 Longer follow-up data presented at the 2022 annual ASCO meeting confirmed the above data.76 Table 1 summarizes the key findings of the pivotal trials investigating EV.77–79

Is there an optimal sequence of treatment in the second line and beyond?
While the results from EV-301 are impressive and show that EV has a superior OS, PFS and ORR compared with single-agent chemotherapy, a remaining question concerns the optimal sequencing of therapies for aUC. EV-301 randomized patients to either EV or investigator-chosen single-agent chemotherapy (taxane or vinflunine).5 Outcomes with single-agent chemotherapy in this setting remain poor, with ORR ranging from around 13% to 30%.80–82 Further, several new agents have been approved in the salvage setting over the last few years for patients who progressed on first-line platinum-based chemotherapy, including ICI (pembrolizumab, avelumab, nivolumab) for patients who did not receive avelumab maintenance, erdafitinib (for those with susceptible FGFR2 or FGFR3 alterations) and sacituzumab govitecan.41–43,48,83 However, there are currently no head-to-head trials investigating response and survival with EV compared with these other approved agents. Consequently, the optimal treatment sequence and when to use EV needs to be determined on a patient-by-patient basis. EV is currently approved for patients who are not eligible to receive cisplatin who have progressed on both platinum-based therapy and ICI and for patients who are not eligible for cisplatin who progress on ≥1 line of therapy. For patients treated with platinum-based chemotherapy followed by switch maintenance avelumab, EV would be a very reasonable subsequent therapy after progression on avelumab. The same is also true for those who receive second-line ICI after progression on platinum-based chemotherapy. However, a retrospective study has suggested that patients with liver and/or bone metastases may have a lower response rate and shorter survival with ICI compared with those without liver and/or bone metastases,84 whereas the same may not be the case with EV.6 Therefore, in selected patients with high-risk features (e.g. visceral/bone metastasis), EV or erdafitinib (for patients with susceptible FGFR2 or FGFR3 alterations) could potentially be considered prior to ICI, in the absence of head-to-head comparisons, while awaiting for the THOR phase III trial and large real-world datasets.85,86
There may also be relevant medical comorbidities that could impact the therapy sequence in aUC. For example, in the EV clinical trials, including EV-301, patients with (sensory or motor) neuropathy grade≥2, uncontrolled diabetes mellitus (defined as glycated haemoglobin ≥8 or between 7 and <8 with symptoms, such as polyuria and polydipsia), active keratitis or corneal ulcerations were excluded, given the concern that treatment-related adverse events could worsen baseline comorbidity. Therefore, if a patient has any of these medical comorbidities, alternative therapies (e.g. sacituzumab govitecan) or enrolling in a clinical trial (e.g. TROPiCS-04) could be considered.58 In addition, in a patient with an FGFR2– or FGFR3-activating mutation or fusion, erdafitinib could be considered prior to or after EV, given the lack of head-to-head comparison between erdafitinib and EV. Although no prospective data are available, a small retrospective study showed that, in patients with FGFR3 alteration, receiving EV prior to erdafitinib was associated with shorter PFS and lower ORR compared with receiving EV after the progression on erdafitinib.87 Another small study showed that none of seven patients with FGFR3 alteration responded to FGFR inhibitor given post EV, while one patient who received FGFR inhibitor prior to EV responded.88 However, larger and prospective studies are needed to validate and inform optimal therapy sequence in aUC.
Use of biomarkers to evaluate response to enfortumab vedotin
Another potential way to further guide therapy selection would be to identify predictive biomarkers. Predictive biomarkers could help identify which patients are likely to benefit more from EV and could further inform treatment decisions, including optimal sequence of therapies.
Nectin-4 expression remains an elusive biomarker, and on-going studies are investigating its efficacy as a reliable predictive biomarker.34,89–91 There seems to be variation between different studies investigating Nectin-4 expression in UC. Nectin-4 expression in tissue is calculated and quantified using the H-score, a histochemical scoring system determined by the staining intensity of the cells (ranging from 0 for no signal to 3 for strong signal) multiplied by the percentage (%) of cells stained at a given intensity. H-scores range from 0 to 300, with a score of 0–99 defined as negative or weak, 100–99 as moderate and a score of 200–300 as strong. In a study of 524 primary bladder tumours, moderate-to-strong H-scores (H-score ≥100) were found in only 63% of samples.69 However, EV-101 and EV-201 revealed that Nectin-4 expression was observed in the vast majority of patients. In the phase I trial EV-101, Nectin-4 expression was initially a requirement for study enrolment; however, almost all of the screened samples exhibited high levels of Nectin-4 expression by immunohistochemistry (IHC), and thus, the protocol was amended, and the requirement was removed.72 Consequently, Nectin-4 expression was also not required for entry in either EV-201 or EV-301. In the EV-201 trial, all 120 patients had detectable Nectin-4 expression with a median H-score of 290.4 Tissue samples were obtained for an exploratory analysis of Nectin-4 expression in EV-301, and results are still pending.5 Possible discrepancies in Nectin-4 staining between EV trials and other studies investigating Nectin-4 expression could be due to the different antibodies used in the IHC testing. Given this variability, IHC expression of Nectin-4 might not be a reliable predictive biomarker for EV response. However, the quantification of Nectin-4 through other methods, such as Nectin-4 mRNA expression (using techniques such as RNAseq or Nanostring transcription), are currently under investigation as potential biomarkers. For example, a retrospective cohort study investigating Nectin-4 mRNA expression in patients with localized bladder tumours across different molecular subtypes found that mRNA expression was heterogeneous across the different subtypes and significantly increased in luminal subtypes.92 Further, in vitro analysis revealed that decreased expression of Nectin-4 in luminal cells led to resistance to EV.92 This may suggest that Nectin-4 RNA and molecular subtyping might be potential biomarkers, though further prospective validation is needed.
Genomic sequencing could also provide insights into potential molecular biomarkers. In a retrospective analysis of next-generation sequencing data of patients with aUC treated with EV, the presence of TP53 and the absence of CDKN2A and CDKN2B alterations were associated with favourable response and improved outcomes, suggesting that these might be potential biomarkers that might predict a response to EV but require further validation.93
Clinical biomarkers may also play a pivotal role in helping inform treatment decisions. The Bellmunt risk factors (Eastern Cooperative Oncology Group performance status <0, liver metastasis, haemoglobin <10 g/dL) were developed based on salvage chemotherapy trials and have been used for clinical prognostication and clinical trial benchmarking.94 In addition, a retrospective study using a large, multinational database of patients with aUC treated with ICI showed that, among patients with aUC treated with second-line ICI after progression after first-line platinum-based chemotherapy, time to second-line ICI <3 months after initiation of first-line chemotherapy was associated with worse outcomes to second-line ICI compared with those who initiated second-line ICI <6 months after initiation of first-line chemotherapy.95 In a separate study using the same database, patients with bone and/or liver metastases in aUC treated with ICI were associated with worse outcomes compared with those without bone and/or liver metastases.84 Perhaps, early progression on previous treatment lines, such as platinum-based chemotherapy and/or ICI, as well as the site of metastases, could also be prognostic biomarkers; however, a predictive role, particularly for EV versus other therapy, remains questionable. Large-scale retrospective and prospective analyses are needed to further investigate and validate both clinical and molecular biomarkers for therapy.
Another useful way translational studies may inform our understanding of EV is through the elucidation of mechanisms of resistance. Putative mechanisms of resistance to EV have not yet been fully understood. In preclinical mouse models with breast cancer, tumours resistant to an EV-like antibody–drug conjugate, comprising a human anti-Nectin-4 monoclonal antibody conjugated to MMAE, named N41mab-vcMMAE, showed increased expression of ABCB1, which encodes the multidrug resistance protein MDR-1/P-glycoprotein.96 The overexpression of this protein has been associated with chemotherapy resistance of tumour cells by preventing the intracellular accumulation of cytotoxic agents.97 Interestingly, the preclinical model showed that the addition of P-glycoprotein pharmacological inhibitors restored the sensitivity of resistant tumour cells to N41mab-vcMMAE.96 Preclinical models have also shown that loss of Nectin-4 expression was also associated with EV resistance.92 On-going studies and biomarker analyses are needed to help further understand mechanisms of resistance to EV and other antibody–drug conjugates.
Looking ahead to the future of enfortumab vedotin: earlier therapy settings?
Given the favourable results of EV in the salvage setting for aUC, considerable enthusiasm remains for exploring whether EV can be moved to earlier therapy lines, either alone or in combination with other therapies. Both the EV-103 and EV-302 trials are investigating response and outcomes with first-line EV alone or in combination with pembrolizumab. EV-103 is a phase Ib/II multicohort, dose-escalation and dose-expansion trial investigating the safety and activity of EV in combination with other agents commonly used in aUC, such as pembrolizumab or cisplatin.77 EV-103 cohort A assessed the safety and activity of a combination of EV and pembrolizumab as first-line therapy in patients with aUC not eligible for cisplatin and showed a manageable safety profile and very promising anti-tumour activity, with a confirmed ORR of 73% (17.8% CR), a median response duration of 25.6 months, an mPFS of 12.3 months, mOS not reached, and a median follow-up time of 24.9 months.77 Early results from a follow-up two-arm cohort K of EV plus pembrolizumab (the other treatment arm was EV alone, but the trial was not powered to compare the two arms) as first-line treatment for patients not eligible for cisplatin with aUC showed an ORR of 64.5% in the combination arm and 45.2% with EV alone, with a few differences in the toxicity profiles.79
EV-302 is a randomized phase III trial investigating a combination of EV plus pembrolizumab versus gemcitabine plus cisplatin or carboplatin (followed by switch-maintenance avelumab, when appropriate, based on a subsequent trial amendment) in previously untreated patients with aUC eligible to receive platinum.34 The primary endpoints of EV-302 are OS and PFS; this trial has not reported data yet. Given the established role of EV after platinum-based chemotherapy and ICI, it will be important to ensure whenever possible that the trial participants in the control (chemotherapy) arm have access to the appropriate off-trial, post-protocol therapies (e.g. ICI, EV) to best inform whether upfront EV/pembrolizumab truly improves OS in this population. Moreover, it will be important to assess the proportion of patients who receive cisplatin as well as the proportion who receive switch maintenance avelumab in the control chemotherapy arm of the EV-302 trial. The question is whether the FDA will provide accelerated approval to EV/pembrolizumab as upfront therapy in patients with aUC not eligible to receive cisplatin based on the results of the EV-103 trial before the EV-302 phase III trial is completed. A future discussion may also include the evaluation of potential dose de-escalation strategies looking at efficacy, safety, quality of life and cost-effectiveness.
In addition, on-going trials are investigating the role of EV, either as monotherapy or in combination with pembrolizumab or other ICI in the perioperative setting.89–91,98 Preclinical data have demonstrated the ability of EV to induce immunogenic cell death and enhance tumour cell killing through the‘bystander effect’.99 Preliminary data from EV-103 cohort H, a single-arm cohort investigating neoadjuvant EV in patients with MIBC who are eligible to receive cisplatin, has shown a pathological downstaging rate of 50% and a pathological CR rate of 36.4%.78 EV-103 cohort L is investigating perioperative EV monotherapy in patients with MIBC not eligible for cisplatin.89 Patients will receive three cycles of neoadjuvant EV followed by radical cystectomy and then six additional cycles of adjuvant EV. The primary endpoint is the pathological CR rate. KEYNOTE-B15/EV-304 (ClinicalTrials.gov identifier: NCT04700124) is a phase III trial investigating perioperative EV plus pembrolizumab versus neoadjuvant chemotherapy with cisplatin/gemcitabine in patients with MIBC who are not eligible to receive cisplatin.90 Patients randomized to receive EV plus pembrolizumab will receive four cycles of neoadjuvant EV plus pembrolizumab followed by five cycles of adjuvant EV and 13 cycles of adjuvant pembrolizumab after radical cystectomy. Primary endpoints are pathological CR rate and event-free survival. KEYNOTE-905/EV-303 (ClinicalTrials.gov identifier: NCT03924895) is a phase III trial investigating a perioperative combination of EV with pembrolizumab versus no experimental perioperative therapy in patients with MIBC who are not eligible for cisplatin.91 Primary endpoints are pathological CR rate and event-free survival. VOLGA (ClinicalTrials.gov Identifier: NCT04960709) is a phase III trial investigating the efficacy and safety of neoadjuvant EV plus durvalumab/tremelimumab versus EV plus durvalumab versus no experimental perioperative therapy in cisplatin-ineligible patients with MIBC.98 These trials are actively enrolling patients and have the potential to change the treatment paradigm of MIBC. Many active, on-going trials are investigating combinatorial strategies with EV in both the perioperative and the aUC setting. Table 2 lists examples of on-going trials investigating EV in this cancer.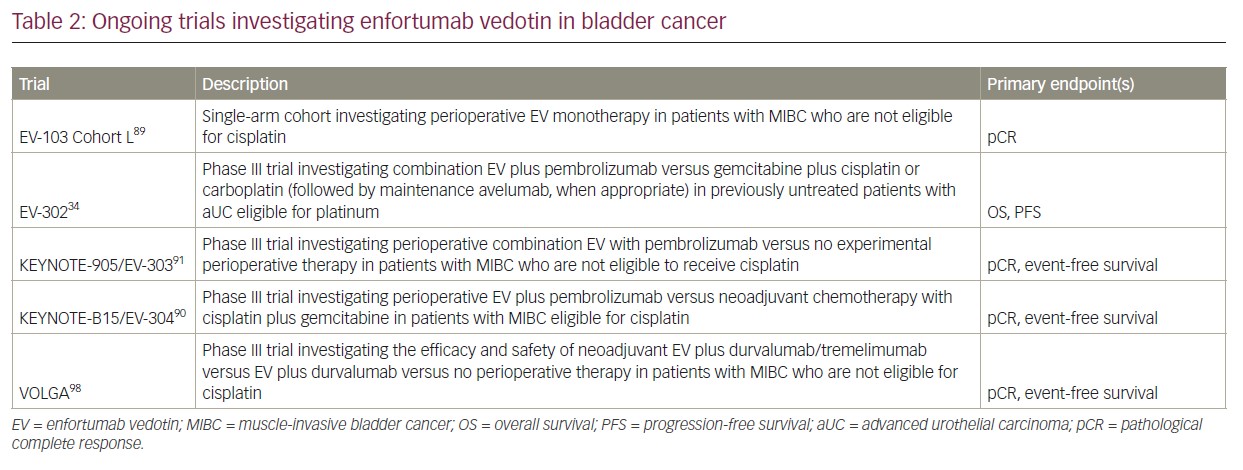
Conclusion
EV is a highly active drug that has already made a very positive impact on aUC. EV-301 was a landmark phase III trial that led to the regular FDA approval of EV in patients who progress after platinum-based therapy and ICI. EV can also be used as a second-line therapy in patients who are not eligible to receive cisplatin. Optimal patient and provider education, recognition and careful management of toxicity by a multidisciplinary team remain essential.74 However, multicentre collaborations, randomized trials with approved agents, promising combinations and biomarker-driven studies can help to determine which patients can benefit more from EV and to elucidate the most appropriate therapy setting and sequence. Moreover, biomarker discovery and validation require translational studies and can inform resistance mechanisms.