Breast cancer is the most common cancer among women worldwide, and more than a fifth of those diagnosed will develop incurable metastatic disease.1 Molecular testing to investigate genetic and genomic variation is essential to identify the most effective treatment plans for patients. Treatment options available for patients with breast cancer are directed through the receptor status – hormone receptor (HR) and human epidermal growth factor receptor 2 (HER2) – with the most common phenotype being HR positive (HR+) HER2 negative (HER2-).2–4
For a long time, single-agent endocrine therapy was the standard of care for first-line treatment in HR+/HER2- metastatic breast cancer.5,6 In 2016, analysis of the PALOMA-2, MONELESSA-2, -3 and -7, and MONARCH-3 trials showed the significant progression-free survival (PFS) benefit in first-line metastatic breast cancer from incorporating cyclin-dependent kinase 4/6 (CDK4/6) inhibitors into endocrine treatment, highlighting the introduction of CDK4/6 inhibitors as truly practice changing.7–16
Following this, the 2020 European Society for Medical Oncology (ESMO) 5th International Consensus Guidelines for Advanced Breast Cancer recommended endocrine therapy and CDK4/6 inhibitors as first-line treatment for HR+/HER2- metastatic breast cancer.6 In the second-line setting, endocrine treatment options include fulvestrant, everolimus-exemestane and alpelisib-fulvestrant.7,17,18 Patients known to be breast cancer gene (BRCA) positive would be eligible for the poly-ADP ribose polymerase inhibitor olaparib.19 Such options provide a heightened opportunity to delay starting chemotherapy, although, for patients who develop endocrine-resistant/refractory disease without such mutations, the only available options are single-agent chemotherapy.6
Anthracyclines and taxanes are the preferred first-line chemotherapy agents for patients with metastatic breast cancer who have not previously received such treatment or have a disease-free interval >12 months following adjuvant treatment.17,18 On progression, single-agent options include capecitabine, vinorelbine, eribulin and platinum-based agents. Due to its effects on patients’ quality of life, single-agent chemotherapy is preferred over combination regimens.17,20 Unfortunately, despite multiple systemic therapy options, many patients continue to experience disease progression while on treatment.21
Sacituzumab govitecan is a novel antibody–drug conjugate (ADC) that has improved both PFS and overall survival (OS) in patients with triple-negative breast cancer (TNBC) after at least two prior lines of treatment.22 Within the HR+/HER2- patient cohort, the phase III trial TROPiCS-02 (ClinicalTrials.gov identifier: NCT03901339) has also shown significant improvement with sacituzumab govitecan in PFS and OS, plus further improvements in the duration of response and overall health-related quality of life.23,24
Antibody–drug conjugates
There is an evolving landscape for the use of ADCs as a novel therapy to improve survival and impinge progression in patients with metastatic cancer. While a particularly recent introduction, the theory of ADCs was first hypothesized in 1910 by Paul Ehrlich as a ‘magic bullet’ that would target cells and deliver treatment.25 Eventually, the development of hybridoma technology in the mid-1970s allowed this theory to become a reality.26 In 2000, the first ADC, gemtuzumab ozogamicin, received US Food and Drug Administration approval for the management of acute myeloid leukaemia.25,27
ADCs target delivery of cytotoxic agents via an antibody. Each ADC comprises a monoclonal antibody (immunoglobulin G [IgG]), a cytotoxic payload, and a chemical linker. The cytotoxic payload is attached to the tail of the antibody via a cleavable linker. The linker’s stability is vital to ensure the conjugate is maintained in the circulation when administered intravenously, and only cleaved after it is identified and internalized by the cancer cell. When a protein is expressed on the surface of specific tumour cells, ADCs deliver the cytotoxic payload upon binding with the antibody target inducing apoptosis of the cancer cell.25
The antibody component of the ADC is designed to target an antigen that is expressed solely or in a significant abundance on tumour cells. The conjugate is internalized or endocytosed upon binding by the tumour cell, developing into an endosome. The endosome becomes a lysosome, creating an environment that breaks the linker and releases the cytotoxic therapy. Typically, this therapy disrupts the host cell’s DNA or microtubules, resulting in apoptosis.25,28,29
An emphasis of the ADC’s theoretical capability is the delivery of anti-cancer therapy directly, therefore, minimizing the significant side effect profile we typically see with standard chemotherapy and subsequently the impact on patients’ quality of life.30 An additional benefit of some ADCs is the bystander effect, where the payload can diffuse into the surrounding cells, resulting in further adjacent cell death.31,32
ADCs are currently in their third generation, with improved linker stability and potency of cytotoxic payload, less off-target toxicity and increased effectiveness against low-prevalent antigens.25,28,29 In managing breast cancer, trastuzumab emtansine (T-DM1) was the first approved ADC for patients with HER2+ metastatic breast cancer after the EMILIA trial showed a benefit in median PFS and OS of trastuzumab emtansine compared with lapatinib and capecitabine. This ADC used the HER2-targeted elements of trastuzumab alongside the delivery of a microtubule-inhibitory agent, DM1.33
With their development, ADCs were designed to increase specificity and therefore minimize the impact of systemic side effects. However, akin to chemotherapy, studies show a maintained presence of haematological toxicity, with bone marrow suppression persisting as a grade 3–4 toxicity for some ADCs.25 Additionally, we must have an increased awareness of unexpected side effects, such as the observed impact of interstitial lung disease following administration of the ADC, T-DM1, which had a significant impact on morbidity and mortality for those affected.34 This is potentially a consequence of the inappropriate uptake of ADCs by, and release of the cytotoxic agent in healthy lung cells. Although the aetiology remains uncertain, prompt identification and management are warranted for all patients.34
Sacituzumab govitecan
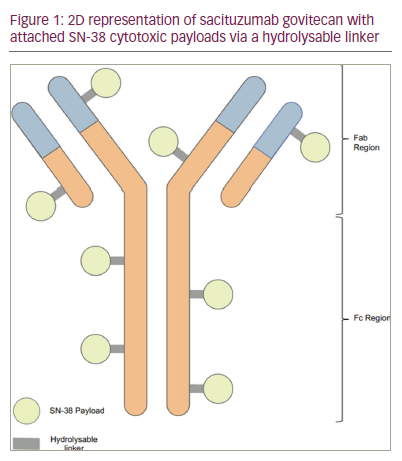
Sacituzumab govitecan is a humanized IgG1κ ADC that targets human trophoblast cell-surface antigens (Trop-2). Trop-2 is a transmembrane glycoprotein encoded by the tumour-associated calcium signal transducer 2 (TACSTD2) gene, often over expressed in epithelial cancers. Typically, Trop-2 is involved in signalling cell migration and anchorage-independent growth31,32,35 Trop-2 is widely expressed across multiple tumour types and is increased in aggressive disease, so it is the ideal target for developing a ‘broad-spectrum’ agent.36
The cytotoxic payload is bound to the Trop-2 antibody via a hydrolysable linker. The linker is released following linkage with Trop-2. SN-38 is membrane permeable when free, providing an extra impact on adjacent tumour cells through the bystander effect, as previously mentioned. SN-38, the active metabolite of irinotecan, works by inhibiting topoisomerase I, thereby preventing DNA replication and transcription and promoting cell death.22,31,32,35,36 A representation of the ADC is reflected in Figure 1.
Development of the TROPiCS-02 trial
Sacituzumab govitecan has clinical benefit in managing patients with relapsed or refractory metastatic TNBC. In the phase III ASCENT trial (ClinicalTrials.gov identifier: NCT02574455), the ADC was shown to significantly prolong PFS by 5.6 months (95 confidence interval [CI] 4.3–6.3; 166 events) versus 1.7 months (95% CI 1.5–2.6; 150 events), hazard ratio (HR) for disease progression or death 0.41 (95% CI 0.32–0.52; <0.001), and OS by 12.1 months (95% CI 10.7–14.0) versus 6.7 months (95% CI 5.8–7.7), HR for death 0.48 (95% CI 0.38–0.59; <0.001) compared with single-agent chemotherapy.22 Subsequently, sacituzumab govitecan has been adopted into the guidelines for the management of TNBC after two or more systemic therapies, at least one for metastatic disease.16
Within the endocrine-resistant/refractory, HR+/HER2- patient population, there is an unmet need for more treatment options, away from chemotherapy, to improve survival and quality of life. Once endocrine resistance develops, current single-agent chemotherapy response rates are low, with multiple studies indicating poor figures for PFS, overall response rate (ORR) and adverse effects.37–39 With this, the innovation in ADCs is a potential area for development.
In 2012, a phase I/II trial (IMMU-132-01; ClinicalTrials.gov identifier: NCT01631552) commenced studying the safety profile and ORR of sacituzumab govitecan in epithelial cancers. In a small population of patients with HR+/HER2- breast cancer patients, a significant improvement in ORR of 31.5% and median PFS of 5.5 months compared with expected values from chemotherapy was identified and gained interest.39 Continuing with this promise and an acceptable safety profile, the multicentre, randomized, open-label phase III study, TROPiCS-02, endeavoured to explore the effectiveness of utilizing sacituzumab govitecan as a novel treatment for patients with endocrine-resistant HR+/HER2- metastatic breast cancer.
TROPiCS-02 study results
This multicentre, global, phase III trial assigned 543 patients to sacituzumab govitecan (10 mg/kg on 1 and 8 of every 21 days) or physician’s choice of chemotherapy (eribulin, capecitabine, gemcitabine or vinorelbine). Patients were heavily pre-treated, with the trial including patients who had progressed or relapsed after 2–4 systemic chemotherapy regimens and at least one hormone treatment and one CDK4/6 inhibitor in the metastatic setting.40 The primary outcome measure was PFS, with secondary measures including ORR, OS, clinical benefit rate, duration of response, safety, tolerability and quality of life. HER2 negativity was defined by an immunohistochemistry score of 0, 1+ or 2+ with a negative fluorescence in situ hybridization (FISH).
The interim analysis for TROPiCS-02 supported the on-going development of sacituzumab govitecan within the endocrine-resistant, HR+, metastatic setting. There was a 34% reduction in risk of progression or death (HR 0.66, [0.53–0.83]; p=0.0003) and a significant median PFS benefit, 5.5 months with sacituzumab govitecan (95% CI 4.2–7.9) versus 4.0 months with chemotherapy (95% CI 3.1–4.4).37 In the updated results presented at ESMO Congress 2022, there was a significant OS benefit of 14.4 versus 11.2 months for those treated with sacituzumab govitecan versus chemotherapy, respectively (HR 0.79, p=0.020), with an additional benefit in health-related quality of life and a delay in deterioration of fatigue.23
The toxicity profile of sacituzumab govitecan was consistent with previous studies, yet, there was a greater burden of grade 3–4 treatment-related adverse events (AEs) compared with chemotherapy.22,37,39,41 The most significant burden included neutropenia (51% versus 38%, respectively) and diarrhoea (9% versus 1%, respectively), both in keeping with the effects of the cytotoxic payload SN-38; these were effectively managed with supportive measures. One death was reported in the sacituzumab govitecan group, secondary to neutropenic colitis.23 It is of note that sacituzumab govitecan did not have any reported AEs of interstitial lung disease, as previously noted with other ADCs.34
Conclusion and future clinical practice
The results of TROPiCS-02 are a promising movement towards improving available treatment options for patients with heavily pre-treated, endocrine-resistant, HR+/HER2- metastatic breast cancer. It also provides us with a further understanding of the effectiveness of ADCs targeting Trop-2. Such findings indicate that sacituzumab govitecan would be an effective alternative to single-agent chemotherapy in patients previously treated with at least two to four systemic regimens, including at least one taxane, one anticancer hormonal treatment, and one CDK4/6 inhibitor, as per standard clinical practice.
However, the significance of this will also be affected by the emerging landscape of the ADC trastuzumab deruxtecan and its role in managing patients with HR+/HER-low (defined as HER2 1+ on immunohistochemistry or HER2 2+, negative on FISH) disease.42 The DESTINY-Breast04 trial studied the impact of trastuzumab deruxtecan on the HER2-low, endocrine-refractory, previously treated population of patients, specifically commenting on results from the HR+ group. Its effectiveness was compared with treatment of physician’s choice, including capecitabine, gemcitabine, eribulin, paclitaxel and nab-paclitaxel. The study reported a significant PFS (10.1 versus 5.4 months, respectively; HR 0.51, 95% CI 0.40–0.64; p<0.0001) and OS (23.9 versus 17.5 months, respectively; HR 0.64, 95% CI 0.48–0.86; p=0.0028) benefit in patients on trastuzumab deruxtecan versus those on chemotherapy in this population with HR+/HER2-low disease.42
The results from DESTINY-Breast04 suggest potential competition for adopting sacituzumab govitecan in patients with endocrine-resistant HR+ disease. However, it is not possible to reliably compare each trial, particularly due to differences in available chemotherapy options and inclusion criteria (TROPiCS-02 patients were heavily pre-treated in comparison to DESTINY-Breast04). Nevertheless, the developing field of trastuzumab deruxtecan in the management of HER2-low disease does raise the question of how both treatment options will be implemented into the current treatment paradigm, as guidelines will eventually aim to reflect the results of both studies.
There is no doubt that introducing sacituzumab govitecan into clinical practice will be a welcome addition to the treatment paradigm of endocrine-resistant, HR+/HER2- breast cancer. Research continues with trials such as GBG102-SASCIA and EVER-132-002 further investigating the contribution that sacituzumab govitecan could have to patients with HR+ breast cancer.