Between 1973 and 1998, the incidence and mortality of melanoma in the US increased at rates higher than those for most other preventable cancers.1 In 2007 alone, there were an estimated 59,940 cases of melanoma diagnosed and 8,110 reported mortalities.2 Therapeutic approaches currently undertaken include cancer-specific vaccines, chemotherapy, biochemotherapy, cytokines, specific immunostimulants, and monoclonal antibodies.3 However, currently there is no therapeutic agent or effective therapy to significantly prolong the survival of metastatic melanoma patients.
Gene Expression Profiling in Melanoma Serial Analysis of Gene Expression
In recent years there has been great progress in understanding the genetics of melanoma and the subsequent therapeutic implications. The sequencing of the human genome has significantly advanced the development and utility of high-throughput gene expression profiling techniques such as microarray technology and serial analysis of gene expression (SAGE). Both of these techniques provide a global overview of the gene expression changes in a cell type or sample, but have different advantages.4
Microarrays are much less tedious, expensive, and time-consuming than SAGE, and nowadays encompass oligos that cover the entire human genome. SAGE does not necessitate a prior knowledge of genes and provides a comprehensive profile of the gene expression of a cell by identifying and tallying transcripts without relying on a ratio comparison. New adaptations to the technique also make it more ‘user-friendly.’ Techniques aimed at minimizing the amount of input RNA such as small amplified RNA-SAGE (SAR-SAGE),5 PCR-SAGE,6 and SAGE-lite7 involve polymerase chain reaction (PCR) amplification of template complementary DNA (cDNA). Other modifications not only aim to find known expressed sequence tags (ESTs) and transcripts, but also attempt to link computational biology and bench science. For example, LongSAGE was developed to experimentally assess whether genes predicted in silico exist in vivo,8 and allows for a high-throughput validation of gene predictions. Modifications to this technique have also improved the amount of input RNA required, as well as resolving issues associated with the concatmaerization of long sequences.9,10 Other sequencing projects in combination with linkage analysis have identified key melanoma genes such as BRAF,11 CDKN2A,12 CDK4,13 and MC1R.14
Microarrays
Microarray chips currently encompass anywhere from 44,000 to 60,000 genes and ESTs, and have evolved considerably over the last 10 years. Seminal studies identified genes and pathways important to melanoma biology, including genes such as Wnt5A15 and RhoC.16 Recent studies have been more focused, trying to identify specific changes in subsets of samples based not simply on tumor stage but also on other factors. For example, Hoek et al. were able to classify melanomas based not only on their metastatic capability but also on their transforming growth factor beta (TGF-β) sensitivity, microphthalmia-associated transcription factor (MITF), and wingless-type MMTV integration site family member 5A (WNT5A) status, and proliferative capacity.17 A recent study has used microarray analysis to identify changes in response to imatinib treatment in melanoma patients undergoing such treatment, and showed an inconsistent pattern of downstream ERK activation.18 Other studies have also examined gene expression changes following therapy, opening up the new and exciting field of individualized pharmacogenomics.19 Studies such as these may start to aid our understanding of how and why certain treatments fail, and to better understand how others work and how to improve them.
Other Gene Expression Profiling Platforms— Comparative Genomic Hybridization, Single-nucleotide Polymorphisms, and Beyond
Finally, the microarray platform has expanded beyond simple gene expression at the RNA level to include techniques such as chromosomal amplification via comparative genomic hybridization (CGH).20 Array CGH is far more sensitive than conventional CGH, and can even detect single-copy changes.21 Another technique that lends itself well to the array platform is the identification of single-nucleotide polymorphims (SNPs), which can be arrayed at a high density. SNP analysis in melanoma has identified polymorphisms in several genes ranging from matrix metalloproteinases (MMPs) to cytokine.22–25 MITF, a key transcription factor in melanocyte biology, has been implicated in melanoma progression in a study that combined SNP analysis with gene expression signatures derived from the NCI60 microarray database.26 More recent discoveries such as the discovery of micro-RNAs (miRNAs) that reside within cells and can act as regulators of gene expression can also be arrayed, and their expression in normal versus cancer cells can be compared. A recent study showed that miRNAs could accurately distinguish between more metastatic and less metastic uveal melanomas, and identified two miRNAs (mir-199 and let-7b) as key discriminators between the two groups.27 These miRNAs may prove to be important markers or targets for the management of melanoma. All of these technologies continue to further our understanding of the biology of melanoma progression and aid us in our discovery of better targets for melanoma therapy.
Signaling Pathways in Melanoma
Although identifying the genetic changes that occur during melanoma progression (or in response to therapy, etc.) is a key first step, the functional analysis of the identified genes is essential. Many of these genetic differences involve proteins that reside within signaling pathways that promote proliferation, differentiation, and apoptosis. In this article, we will outline the major pathways that signal to promote melanoma progression, and discuss the components of these pathways and their potential as molecular targets (see Figure 1). These pathways include the WNT5A pathway,28 the TGF-β family,29,30 the mitogen-activated protein kinase (MAPK) pathway,31 the cyclic AMP (cAMP) pathway,32 the Rho/Rac pathway,16 and the phosphatidylinositol 3-kinase (PI3K) pathway.32
WNTs signal through three distinct pathways: the canonical WNT/β- catenin pathway, the WNT/Ca2+ pathway, or the planer cell polarity (PCP) pathway.33 The WNT family of secreted proteins consists of 19 structurally related molecules. Receptors for the WNT pathways include the frizzled family of receptors, LRP5/6, derailed, and Ror2, and the combination of WNT protein and receptor determines which pathway is activated.33 The canonical Wnt pathway is very well understood, and the role of Wnts such as Wnt1 and 3A, and the key mediator of this pathway, β-catenin, has been the focus of intense study for the last decade. However, its role in melanoma remains somewhat controversial. β-catenin is clearly important in the beginning stages of melanoma, initiating melanocyte transformation and tumor growth.34 However, its role in the later stages of melanoma progression is unclear.33
Over the last few years, the non-canonical Wnt pathway, represented by Wnts such as Wnt5A, has begun to receive some attention. It was discovered that increased expression of WNT5A correlates with increased tumor grade, metastatic potential, motility, and invasiveness.28 This provides an attractive target for the modulation of WNT5A-induced signal transduction. Signaling through WNT5A leads to the upregulation of phosphodiesterase (PDE), inhibiting cyclic guanosine monophosphate (cGMP) activity. Subsequently, the Gβ/γ subunits activate phospholipase C (PLC), resulting in the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG).35 DAG activates protein kinase C (PKC), IP3 increases the Ca2+ levels and activates calcineurin and Ca2+ calmodulin- dependent protein kinase II (CamKII).36,37 In addition, it has also been shown that increased expression of WNT5A increases the expression of genes involved in the epithielial to mesychymal transition (EMT), such as vimentin, and also increases metastasis related, genes such as CD44, which is involved in tumor cell homing during metastasis. Furthermore, WNT5A decreases the expression of genes such as KISS-1, a metastasis suppressor.38
In contrast to WNT5A, TGF-β signaling has been shown to promote growth inhibition in ME15 and D10 melanoma cell lines.39 The superfamily consists of two distinct pathways: the TGF-β/activin/nodal and the BMP/GDFs.40 As melanoma progresses, cells escape TGF-β-mediated growth inhibition by expressing the oncogenic proteins Ski and Sno. Ski is a Smad-dependent co-repressor of TGF-β, BMP, and activin.41–43 The inhibition of Ski using RNA interference restores TGF-β-induced growth inhibition.44
The MAPK pathway has also been shown to be important in the progression of melanoma. Stimulation of the majority of growth factor receptors leads to the activation of the MAPK pathway. It is pivotal in controlling cell growth, differentiation, and survival.31 Activation of the extracellular regulated kinase (ERK)/MAPK complex (also known as MEK) occurs through the activation of small GTPases of the Ras family by receptor tyrosine kinases (RTKs).45 MEK can then activate ERK, which subsequently regulates gene expression, metabolism, and cytoskeletal rearrangement,46 an important event in increasing invasiveness and metastatic potential.
Another important pathway in melanoma is PI3K. This is an effector of Ras that converts phosphatidylinositol4,5 phosphate into phosphatidylinositol3–5 phosphate (PIP3), which in turn regulates the activity of pleckstrin homology (PH) domain-containing proteins.47 One very important PH domaincontaining protein activated by PIP3 is protein kinase B, also known as Akt. Among the many substrates for Akt are glycogen synthase kinase 3 (GSK3) and cAMP-response element binding protein.48 This illustrates the importance of signaling pathway cross-talk. GSK3 activity is inhibited by WNT signaling and the activation of GSK3 will phosphorylate β catenin, promoting its degradation. There is some correlation between increased levels of phosphorylated β-catenin and the progression of melanoma,49 and β-catenin expression at the cell membrane is also lost.50 In addition, the activation of Akt by Wnt3A has been shown to increase growth and proliferation in a β-catenin-independent manner,51 possibly through activation of the WNT/Ca2+ pathway.
Finally, other genes known to be involved in melanocyte development and differentiation have been identified by gene expression profiling to be potential melanoma oncogenes. The master regulator of differentiation of the melanocyte lineage, MITF, is activated by the serine/threonine kinase, protein kinase A (PKA), which, in turn, is activated by cAMP.52 This activation can be regulated by the A3 adenosine receptor A3AR. This receptor suppresses the formation of cAMP and promotes growth inhibition by the downregulation of PKA and Akt.53 The amplification of MITF circumvents this regulation, and MITF is able to transform primary human melanocytes when expressed in conjunction with a V600EB-Raf mutant.26 The overexpression of MITF also has implications for the ability of tumors to evade immune surveillance as it is the key transcription factor responsible for the transcription of melanoma differentiation antigens such as MART-1 and GP100.52 These antigens are recognized by tumor-infiltrating T cells, and are often used as targets for immunotherapy. Below, we discuss these and other molecular targets and their potential in melanoma therapy.
Melanoma Therapy
Molecular Targets
Wnt5A can activate PKC, resulting in pleiotropic downstream effects. This indicates that blocking PKC may have beneficial effects, and indeed this enzyme has been the focus of some attention as a molecular target for cancer therapy. In melanoma cells, treatment with rWNT5A increases motility and invasive potential, an effect that can be blocked by using PKC inhibitors.38 Similarly, studies have shown that treating human endothelial circulating progenitor cells (CPCs) with WNT11 (a ligand that also activates the non-canonical WNT/Ca2+ pathway) significantly enhances the differentiation of CPCs to cardiomyocytes and that treatment with the PKC inhibitors bisindolylmaleimide I and III blocked differentiation.54 The PKC inhibitor 7-hydroxystaurosporine has been shown to inhibit tumor cell invasion and promote cell death.55,56 Furthermore, H7, another PKC inhibitor, was shown to decrease tumor cell invasion and metastasis in the melanoma cell line B16-BL6. The expression of MMP-1, -2, -9 messenger RNA (mRNA) and the phosphorylation of the MAPK pathway member, extracellular signal-regulated kinase 1/2 (ERK1/2), also decreased. In addition, similar effects were seen when using U0126, a MEK 1/2 inhibitor, suggesting that H7 inhibits signal transduction through the PKC/MEK/ERK pathway.57 In addition, in B16 cells pre-treatment with H7 reduced the phosphorylation of MEK and promoted apoptosis.58 However, because of the ubiquitous nature of PKC expression, targeting this enzyme directly may prove to be either toxic, or, on the other hand, ineffective due to redundancy in signal transduction pathways.
Another target associated with the MAPK pathway but downstream of the PI3K/Akt pathway is mammalian target of rapamycin (MTOR).59 An inhibitor of MTOR, temsirolimus (CCI-779) is currently in clinical evaluation. Initial studies showed a survival advantage for temsirolimus but later trials demonstrated no single agent activity.60,61 This is also true for inhibitors of one of the most exciting discoveries in recent melanoma research: the B-raf mutant V600EB-Raf. Mutations in the Ras/Raf pathway are seen in over 80% of melanomas and also nevi, and the V600EB-Raf mutant is the most common of the somatic mutations seen.11,62,63 The utility of B-raf inhibitors such as sorefanib is still under investigation. In clinical trials, sorafenib has shown very little benefit when used on its own.64 However, when used in combination with carboplatin and paclitaxel, the agents demonstrated an 85% disease control rate.65 Unfortunately, these agents do have a considerable toxicity.
Immunotherapy
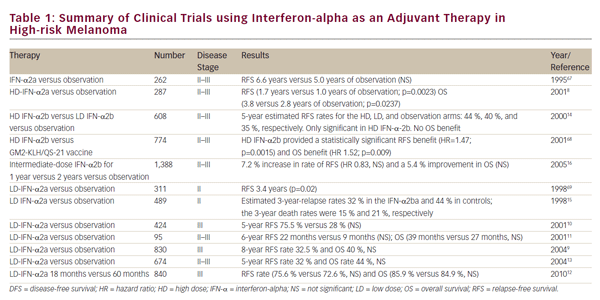
Immunotherapy has long been recognized as a potential treatment for melanoma and an alternative for treating tumors that develop resistance to chemotherapy and radiotherapy. In the past, interleukin (IL)-2 and interferon alpha (IFNα) have been the therapies of choice for patients with resecting stage III melanoma.66,67 However, anticytotoxic T-lymphocyte associated antigen 4 (CTLA-4) antibodies now present a promising new class of treatment. CP-675.206 and ipilumumab (MDX-010) are two different anti-CTLA-4 monoclonal antibodies currently in phase I and II clinical trials.68,69 Both agents are showing promising results with similar efficacy and an objective response rate of 15%, with complete responders at 5%.70 In addition, melanoma differentiation antigens such as MART-1, gp100, and tyrosinase been used as the targets in a multiepitope peptide vaccine.71,72 The vaccine appears to be well tolerated and may be associated withm immunological responses in melanoma.3 Novel techniques to boost the amount of T cells recognizing these epitopes have also been developed,73 and have had some success in patients.74 Unfortunately, for the most part melanoma vaccines have not proved to be very effective in trials to date.70,75
This may be due to the complex and laborious nature of both virus production and delivery. However, gene-therapy-based vaccine approaches using plasmid or viral DNA delivery systems have had some promise for certain targets, including GM-CSF.76 In addition, recent unpublished data from our laboratory indicate that more metastatic cells may lose expression of these antigens, and suggest that the adjuvant use of therapies to inhibit regulators of these melanocyte differentiation antigens may improve the efficacy of these vaccines. Whether vaccine therapy is practical for routine clinical use remains to be seen, but data suggest that there is hope for these therapies, especially when used adjuvant to either conventional or targeted chemotherapy.
Conclusion
As demonstrated throughout this review, melanoma therapy using downstream signal transduction proteins as targets is still in its early days. Progress has been made in areas such as chemotherapy and immunotherapy, but with such complex overlapping signaling networks finding an effective individual target has proven difficult. However, the continuing advances in gene therapy, signal transduction inhibitors, monoclonal antibodies, and immunotherapy, along with the discovery of new molecular targets and our understanding of how those targets are expressed, provide an exciting new route of investigation that will hopefully lead to more effective treatment regimes for melanoma patients. ■
Trending Topic

Trending Topic
Developed by Touch
Mark CompleteCompleted
BookmarkBookmarked
Despite being considered a rare type of malignancy, constituting only 3% of all gastrointestinal cancers, the incidence of biliary tract cancers (BTCs) has been increasing worldwide in recent years, with about 20,000 new cases annually only in the USA.1–3 These cancers arise from the biliary epithelium of the small ducts in the periphery of the liver […]