Autoimmune diseases comprise more than 80 chronic conditions that collectively affect approximately 5 to 8 % of the US population and are a leading cause of death in young and middle-aged women.1 Moreover, the incidence and prevalence of autoimmune diseases are rising. The age of onset of autoimmune diseases varies widely but many start during childhood2 and, being chronic and debilitating in nature, require long-term therapy and invoke considerable medical costs, long-term impaired quality of life, and constitute a significant burden to families and society. Type 1 diabetes (T1D), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), and inflammatory bowel disease (IBD) account for the majority of the patients with autoimmune diseases. Understanding the dysregulated immune response underlying autoimmune diseases will help the development of disease-specific therapeutics. In this short review we will present an overview of the pathophysiology of autoimmune diseases in the context of regulatory T cell (Treg) dysfunction with a focus on the emerging role for interleukin-2 (IL-2) based Treg therapeutics in restoring immune regulation and mitigating organ damage.
Pathophysiology of Autoimmune Disease— The Role of Tregs
Autoimmune diseases are characterized by a breakdown of mechanisms that allow the immune system to distinguish between self and nonself and maintain immunologic self-tolerance. Organ damage and the ensuing clinical manifestations may result from the action of autoantibodies, self-reactive effector T cell (Teff) responses, secreted cytokines, or other elements that participate in the autoimmune response.3 In recent years, there has been increasing interest in the role of Tregs, which are important in the maintenance of peripheral immune tolerance.4 Tregs can suppress immunemediated inflammation through a number of complementary mechanisms that may involve cell–cell contact and the release of regulatory cytokines that directly limit the responses of effector immune cells.
Several subtypes of Tregs exist, the most well studied being CD4+ cells that express high-level CD25 and the transcription factor forkhead box P3 (FOXP3), which is a critical determinant of Treg cell phenotype and function. Treg deficiency or dysfunction is associated with autoimmune disease. For example, in experimental animal studies, genetic or pharmacologic depletion of Tregs causes autoimmunity.5,6
Mutations of the FOXP3 gene in humans result in impaired Treg function and are associated with the ‘immunodysregulation polyendocrinopathy enteropathy X-linked’ (IPEX) syndrome, a rare disorder characterized by fulminant multi-organ autoimmunity.7 In clinical studies, decreased levels of circulating CD25+CD4+ T cells have been reported in patients with autoimmune disease, including vasculitis, rheumatic disease, juvenile idiopathic arthritis, and Kawasaki disease,8–12 and are associated with high disease activity or poor prognosis.8,10,12 Graft versus host disease (GvHD), which is a manifestation of allo-immunity following hematopoietic stem cell transplantation (HSCT), has also been associated with Treg cell deficiency.
These data have led to the hypothesis that augmentation of Tregs may be a useful therapeutic strategy in autoimmune disease. Treg augmentation has resulted in clinical improvements in numerous animal models of autoimmune diseases.13 Furthermore, the administration of in vitro expanded CD4+CD25highCD127-Tregs has been found to be safe and may help to preserve β-cell function in children with T1D.14,15 Targeting Treg cell enhancement at the cellular and molecular levels may therefore be an attractive therapeutic strategy. Cytokines that can modulate and boost Treg-mediated suppression of immune responses may play an important role in the control of autoimmunity. This article will focus on the ability of IL-2 to augment the numbers and function of CD4+ Tregs.
The Role of IL-2 in the Pathophysiology of Autoimmune Disease
IL-2 was originally discovered as a growth factor for activated T cells in vitro. Physiologically, however, IL-2 is essential to maintain the peripheral homeostasis of CD4+CD25highFOXP3+ Tregs. IL-2 and IL-2 receptor (IL-2R) knockout mice have preserved cytotoxic immune responses, but exhibit autoimmunity. In a study in which the genes for IL-2 were inactivated by gene targeting, the knockout mice died early due to lymphoproliferative syndrome accompanied by severe autoimmunity.16 Neutralization of circulating IL-2 by anti-IL-2 monoclonal antibody has also been found to result in a number of autoimmune diseases, including gastritis, thyroiditis, sialadenitis, and severe neuropathy.17 Autoimmunity associated with impaired IL-2 or IL-2R signaling is due to a failure in the development and homeostasis of Tregs.18 Importantly, a low level of IL-2 receptor signaling is sufficient to provide these key functions to Tregs, while not supporting IL- 2-dependent T effector activity. These latter observations provided some of the initial rationale for low-dose IL-2 therapy for autoimmunity.19
CD4+ Tregs do not produce IL-2 but their survival and function depend on IL-2 uptake via the constitutively expressed high affinity heterotrimeric IL-2 receptor complex (CD25, CD122, and CD132).20,21 By contrast, conventional CD4+ and CD8+ T cells constitutively express a moderate affinity IL-2 receptor complex (CD122 and CD132) and do not express CD25; they do not express the high affinity IL-2R complex until they have been activated through their antigen (Ag) receptor. When conventional CD4+ and CD8+ T cells are activated, lL-2 also promotes Th1 and Th2 development while it actively opposes the development of Th17 and Tfh cells.22,23 This dual role for IL-2 in tolerance and immunity suggests that it may have pleotropic effects in the context of autoimmune diseases. As IL-2 is vital for amplifying T-cell-mediated effector immune responses, including T-cell memory, it could function to enhance autoreactive T cells at higher doses. However, the capacity of IL-2 at low doses to support Tregs indicates that it also critically serves as a negative regulator of autoimmunity.6,16 Low-dose IL-2 is defined operationally, i.e. IL-2 doses that preferentially enhance the proliferation, activation, and survival of Treg (versus conventional CD4+ T cells [Tcon]). Practically, the doses used are suitable for outpatient administration. IL-2 is also well known to be important for activation-induced cell death (AICD), which controls the expansion of immune effector cells.24,25 In autoimmune disease e.g. SLE, AICD is decreased and this defect has been considered responsible for the poor elimination of self-reactive T cells.26 Therefore, IL-2 not only promotes effector immune responses but also acts to eliminate activated T cells.
Defects in the IL-2 pathway or reduced IL-2 availability are seen in some autoimmune conditions including SLE27 and T1D,28–30 whereas in others IL-2 deficits are associated with disease progression; these include RA31 and IBD.32 Studies in SLE patients have noted dysregulation of host immune responses, including decreased CD4+ Treg function and/or numbers, decreased AICD and compromised cytotoxic cell responses.27,33–36 The molecular mechanisms responsible for the decreased IL-2 production involve an imbalance between the transcription factors cyclic adenosine monophosphate (cAMP) response element-binding protein (CRE)-binding protein (CREB) and its modulator (cAMP response element modulator-α [CREMα]25,37,38 T cells from patients with SLE express increased amounts of calcium/calmodulin-dependent protein kinase IV (CaMK4) in the nucleus, which phosphorylates CREM to bind to the IL-2 promoter.35 At the same time, T cells express increased amounts of serine/threonine-specific protein phosphatase 2A (PP-2A), which dephosphorylates phosphorylated cAMP response element binding protein (pCREB), tipping the balance of CREB/ CREM toward CREM and suppression of IL-2 promoter activity (see Figure 1).33,39 Recent studies have indicated that estrogen enhances the expression of CREMα, resulting in IL-2 suppression in human T lymphocytes.40 Altered immune function leads to increased rates of infection and tissue inflammation in patients with SLE.41
The mechanisms by which low-dose IL-2 treatment can re-establish immune tolerance have recently been partly elucidated in a study of patients with chronic GvHD, a common complication of allogeneic HSCT arising due to donor effector immune responses to allogeneic (donor/ recipient polymorphic) and autologous (donor/recipient nonpolymorphic) Ags. Chronic GvHD is characterized clinically by multisystem immune inflammation and fibrosis. Immunologically, there is a relative failure of Treg compared with Teff reconstitution after allogeneic HSCT in adults, arising due to reduced CD4+ Treg thymogenesis, with a compensatory peripheral Treg proliferation that ultimately cannot be sustained owing to increased apoptosis and impaired survival of the circulating CD4+ memory Treg. Additionally chronic GvHD patients have a documented imbalance in the activation of CD4+ Treg versus Teff cell activation as measured by phosphorylation of signal transducer and activator of transcription 5 (pStat5) that acts downstream of IL-2 receptor activation on immune cells.20 In GvHD, low-dose IL-2 therapy increased pStat5 activation in Tregs and decreased production of phosphorylated pStat5 in conventional CD4+ T cells. However, this was not a study of patients with T1D42 (Alberton Pugliese, Tom Malek, Michelle Rosenzwajg, and David Klatzman, unpublished data). Treatment with low doses of IL-2 also resulted in restoration of Treg homeostasis, including increased peripheral proliferation, increased thymic neogenesis, and enhanced resistance to apoptosis.43
To summarize, at high doses, IL-2 activates immune Teffs; but at lower doses, IL-2 induces Tregs and also natural killer (NK) cell numbers and function to a lesser extent and in a dose-dependent manner.18 However, it is not known whether the therapeutic effects of low-dose IL-2 are solely attributable to the expansion of Tregs; in an animal model of experimental autoimmune encephalomyelitis (EAE), the IL-2 induced expansion of NK cells contributed to disease protection.44 Overall, these data provide a strong rationale for the therapeutic use of low-dose IL-2 in the treatment of autoimmune inflammation.
Therapeutic Use of IL-2 in Autoimmune Disease
High doses of IL-2 induce proliferation of cytotoxic CD4+ Teff cells, and this led to the use at high doses (100–150 x 106 IU/m2/day) to boost cytotoxic immune responses in patients with advanced cancers. Treatment with high-dose IL-2 led to a durable and complete tumor regression in ~8 % of patients with renal carcinoma and melanoma, leading to its approval by the US Food and Drug administration (FDA) to treat these malignancies. However, administration of high-dose IL-2 was associated with significant toxicity including the development of the vascular or capillary leak syndrome (VLS/CLS).45 At lower doses, IL-2 has been used to boost immune responses in patients with advanced HIV and induced a significant rise in the CD4+ T-cell count compared with patients treated with antiretroviral therapy alone.46
Lowering the dose to reduce toxicity greatly reduces therapeutic efficacy as an immune stimulant. The limited efficacy of high doses of IL-2 in the cancer and HIV setting may also be explained by the unintended, concomitant activation of Tregs, which can suppress the effector responses that the therapy aimed to stimulate.47,48 IL-2 administration increases CD4+ Tregs in cancer patients.48,49 Such increases, while detrimental to IL-2-mediated anticancer therapy, may be advantageous in autoimmune diseases. At high doses, IL-2 activates immune Teffs; however, at low doses, IL-2 induces Tregs and NK cell numbers and function.18 These findings and the high sensitivity of Tregs for IL-2 support the therapeutic use of low-dose IL-2 for autoimmune disease.50
Preclinical Studies
Mouse studies cannot help in determining appropriate dosing of IL-2 as Treg activation by IL-2 in mice require doses in the range of 25–50 x 103 U/ day, which correspond to high doses in humans. IL-2 has been found to have a proliferative effect on Tregs in low doses (0.5–1 × 106 IU/m2/day) in nonhuman primates.51 Administration of IL-2 to lupus-prone mice protected against end-organ damage and suppressed inflammation by limiting the production of IL-17-producing double-negative T cells and by expanding Tregs.52 Interestingly, in this study it was demonstrated that IL-2 acts on CD8+ cells and augments their ability to kill CD3+CD4-CD8- (double negative) T cells,52 which are expanded in SLE patients and lupus-prone mice and contribute to the immunopathogenesis of the disease.36 IL-2 administered to mice with EAE, prior to disease onset resulted in significant improvement in the clinical manifestations of EAE as well as expansion of Tregs while preventing the induction of T helper 17 (Th17) cells.53,54However, the efficacy of low-dose IL-2 in these studies is dependent on disease stage; administration of IL-2 after the onset of clinical symptoms failed to suppress disease despite the induction of Tregs.
In the nonobese diabetic (NOD) mouse model of T1D, low-dose IL-2 protects against disease development55 and reverses established T1D.56 Adeno-associated virus (AAV)-based expression of IL-2 selectively in pancreatic beta cells of NOD mice expanded islet-resident FOXP3(+)Tregs, resulting in effective long-term suppression of T1D.57 Administration of NOD mice with a vector able to deliver IL-2 in an inducible manner resulted in enhanced Treg function and disease improvement.58 In addition, beta cells express the heterotrimeric high affinity IL-2 receptor and there is initial evidence that IL-2 may promote beta cell replication.59,60
Clinical Studies
Studies reported in 1992 and 1994 administered low-dose IL-2 after autologous or T-cell depleted allogeneic stem cell transplantation. Patients received low-dose IL-2 by continuous intravenous infusion for a 90-day period beginning approximately 3 months after transplant. Treatment was well tolerated and the major immunologic effect observed at that time was a marked increase in the number of circulating NK cells.61,62Subsequent examination of cryopreserved blood samples obtained from patients on these studies demonstrated that CD4+CD25+FOXP3+ cells were also expanded during low-dose IL-2 therapy.63 Due to the high toxicity of highdose IL-2, many groups attempted to lower IL-2 doses in the fields of cancer and chronic infection. For example, a phase I study found that administration of low-dose IL-2 (0.9 × 106 IU/m2/day for 8 weeks) is safe and relatively well tolerated in patients with HIV infection or HIV and cancer.64
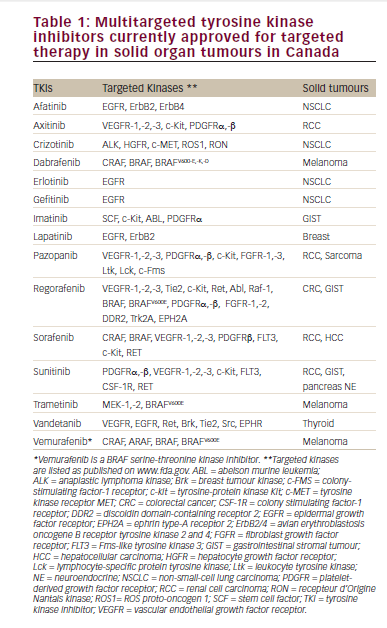
Subsequent clinical studies investigating the use of low-dose IL-2 in autoimmune diseases are summarized in Table 1. Two successful clinical trials of low-dose IL-2 in the treatment of auto and allo-immune diseases have been reported. Patients (n=10) with hepatitis C-induced vasculitis that was refractory to standard antiviral therapy, rituximab or both, were given low-dose IL-2 (1.5 × 106 IU once a day for 5 days followed by 3 × 106 IU for 5 days for 3 cycles on weeks 3, 6, and 9).65 Percentages of Tregs constantly and significantly increased at each course. By week 9, there was a significant increase in the proportion of CD4+ Tregs, from 3.6 % at baseline to 11.8 % (p=0.004). The selective expansion of Tregs was associated with marked clinical improvement, and the treatment did not induce Teff activation or vasculitis flare, further demonstrating the selective effect of low-dose IL-2 on Tregs and no Teffs in vivo. The treatment also did not increase HCV viremia. There were no significant changes in circulating levels of granulocytes, red cells, or liver enzymes and no adverse events (AEs) related to the treatment higher than grade 1. A clinical improvement of vasculitis was seen in eight of 10 patients.
Another study investigated the use of IL-2 in patients with chronic GvHD (cGvHD). Patients (n=29) with cGvHD refractory to glucocorticoids were given daily low-dose IL-2 treatment (either 0.3 × 106, 1 × 106, or 3 x 106 IU/ m2/day) for 8 weeks.66 The highest dose, however, resulted in unacceptable grade 2 AEs, including fever, malaise, and arthralgia. Thus, the maximum tolerated dose was 1 × 106 IU/m2/day. Low-dose IL-2 treatment had an acceptable toxicity profile and did not induce significant leukopenia, neutropenia, thrombocytopenia, or hepatic dysfunction. Of the 23 patients who could be evaluated for a clinical response, 12 had a partial response involving multiple sites and 11 had stable disease. No patient had cGvHD progression. Immunologically, the peak median number of CD4+ Tregs at 4 weeks was more than eight times higher than at baseline (p<0.001), without any change in the CD4+ Teff cells. The NK cell count also rose above baseline. Additionally, daily low-dose IL-2 resulted in restoration of various parameters of Treg homeostasis, including peripheral proliferation, thymic neogenesis, and enhanced resistance to apoptosis.43 Low-dose IL-2 therapy also preferentially increased pStat5 activation status of Tregs, to resolve the CD4+ Treg:Teff pStat5 activation imbalance noted in cGvHD patients. Extended low-dose IL-2 therapy led to sustained clinical and immunologic responses (see Figure 2), enabling tapering of glucocorticoids by an average of 60 %. However, despite Treg-cell expansion, clinical benefit was not seen in all patients. Clinical response may be influenced by numerous factors, including patient characteristics, underlying disease, concomitant immunosuppressive medications, and the extent of tissue injury. Treg-cell activity may also vary from one patient to another. Clinical response rate was higher in patients with an above median Treg/Teff ratio at study entry. Further studies are required to confirm these findings.
In T1D, in a recent phase I/II study, 24 adult patients with T1D were randomized to three IL-2 dosing regimens. This was the first randomized placebo-controlled, double-blind clinical study of low-dose IL-2 in an autoimmune disease. IL-2 was well tolerated at all doses, with no serious AEs. Nonserious AEs, the most common of which were injection-site reaction and influenza-like syndrome, were slightly more frequent in the higher-dose group. Treatment with IL-2 increased in the proportion of Tregs at all doses compared with placebo (see Figure 3).42 While the study was not powered to detect significant improvements of insulin secretion, no impairment was observed. At the 1 × 106 U/day dose, there were no elevations of Teffs or of NK cells, suggesting that this dose may be both efficacious and safe. Thus, there is no evidence so far that low-dose IL-2 may worsen beta cell function as a result of direct effects on beta cells or activation of autoreactive effector cells.
Alopecia areata is an autoimmune disease associated with infiltration of CD4+ and CD8+ T cells around the hair follicles. In a prospective pilot study, five patients with alopecia areata were treated with low-dose IL-2. Of these, four patients attained considerable hair regrowth, as well as an increase in Tregs and no serious AEs. Histologic examination revealed a reduction of infiltrating CD8+ T cells paralleled by an increase of FOXP3 Tregs.67 A phase I/II study in patients with severe and resistant alopecia areata is currently recruiting.
Further clinical studies are in progress, including the phase II Induction of Tregs by low-dose IL-2 in Autoimmune and Inflammatory Diseases (TRANSREG) study that will assess the safety and biological efficacy of low-dose IL-2 as a Treg inducer in 11 autoimmune and auto-inflammatory diseases: RA, ankylosing spondylitis, SLE, psoriasis, Behcet’s disease, Wegener’s granulomatosis, Takayasu’s disease, Crohn’s disease, ulcerative colitis, autoimmune hepatitis, and sclerosing cholangitis.68
A 9-week, single-center, nonrandomized, single-dose, open label, adaptive dose-finding trial, the Adaptive study of IL-2 dose on Tregs in T1D (DILT1D), is ongoing.69 The ultra-low-dose IL-2 to fight T1D trial (DIABIL-2), a doubleblind, randomized, European phase IIb clinical trial, is also in progress.70
In addition, ultra-low-dose IL-2 (0.1 or 0.2 × 106 IU/m2/day) was administered to 12 healthy volunteers with minimal AEs, mainly grade 1–2 injection site reactions. The levels of CD4+ Treg and CD56bright NK significantly increased at 7 days.71 Low-dose IL-2 may affect the function of additional immune cells including innate lymphoid cells 2 (ILC2), which express CD25.72 Finally, a recent case study has reported that low-dose IL-2-therapy induced a rapid, strong, and sustained reduction of disease activity as well a substantial and selective expansion of the Treg population in a SLE patient with increased disease activity.73 Further cases of SLE improvements after low-dose IL-2 have also been recently reported.74,75
Practical Aspects of Administration of Low-dose IL-2
Patients with autoimmune diseases represent a heterogeneous group and it may be difficult to predict an optimal IL-2 dosage a priori and this may also vary by underlying condition. Thus, caution must be exercised in translating IL-2-based therapies to the clinic. Factors to consider include the definition of an appropriately low dose of IL-2 that is efficacious and safe in a specific autoimmune disorder, also the consideration of the frequency of administration and the cumulative dosing as a function of treatment duration. The potential effects of concomitant therapies must also be considered, if given in combination. Future trials should further evaluate individual patient’s responses versus dosing and frequency of administration, and aim at developing biomarkers of IL-2 response that could be used to personalize doses, especially in the context of combinatorial therapies. For instance, a study of low-dose IL-2 to enhance CD4+ T cell counts in patients with HIV found that daily dosing was associated with fewer adverse effects than intermittent dosing.76 Furthermore, IL-2 has a very short half-life in human serum (5–7 minutes).77 Subcutaneous or intraperitoneal injections can prolong the half-life.78 In addition, coupling IL-2 to IL-2 monoclonal antibodies prolongs its half-life, leading to a 40- fold higher in vivo biological activity compared with soluble IL-2, and may offer a promising approach to the treatment of autoimmune disease.79 The efficacy of this approach has been demonstrated in experimental models of the autoimmune disease myasthenia gravis.80 Attempts to couple IL-2 to larger proteins, such as albumin and unrelated antibodies to create IL-2- IgG fusion proteins, have had limited success.20
Use of Low-dose IL-2 in Conjunction with Other Therapies
Another approach to the treatment of autoimmune disease with low-dose IL-2 is in combination with immunosuppressive drugs that are selectively sparing of Treg. In T1D patients, IL-2 was first tested in combination with rapamycin, a drug that blocks cell-cycle progression and cytokine signal transduction through inhibition of the mammalian target of rapamycin (mTOR), another downstream IL-2 signaling component. Rapamycin increases the production of Tregs and also enhances their suppressive ability.81,82 The combination of IL-2 and Rapamycin has been shown to promote induction, survival, and expansion of functional Treg from CD4+CD25- cells.83 Moreover, combined therapy with IL-2 and rapamycin prevents T1D in animal models.84 However, a phase I clinical trial involving combination therapy with IL-2 and rapamycin reported a temporary worsening of C-peptide production. In this study, nine adult patients were treated with rapamycin for 3 months.85 During the first month, patients were also treated with 12 injections of IL-2, given at a dose of 4.5 × 106 IU, 3 times a week. Analysis of the 3-month data for seven patients revealed that they all had experienced a transitory but significant reduction in stimulated C-peptide responses. It is important to consider that IL-2 was given only during the first month, and yet the impaired secretory was observed at 3 months, while patients were still treated with rapamycin but were no longer treated with IL-2. Since patients were not evaluated earlier during the IL-2 course, the actual effects of this combination could not be directly evaluated. Insulin secretion improved after rapamycin was discontinued. This should not be surprising, since rapamycin is known to have a negative impact on beta cell function, survival, and peripheral insulin resistance; rapamycin inhibits the mTOR pathways, causing beta cell toxicity, reduced beta cell size, mass, proliferation, impaired insulin secretion, increased apoptosis, autophagy, and peripheral insulin resistance.85–87 Thus, at least in the T1D setting, the effects of rapamycin on pancreatic beta cell function may overcome any possible synergistic effect on Tregs when given with low-dose IL-2.
Adoptive cell therapy involving the combination of IL-2 and infusion of Tregs is also the subject of clinical investigations, although research is at an early stage. This approach was shown to expand Tregs in vivo following allogeneic HSCT.88 Translation of these promising data to clinical practice will require a robust method of generating large pure populations of Agspecific Tregs; an alternative is the use of ex vivo expanded Tregs89 or freshly isolated donor Tregs.90
Further clinical development of low-dose IL-2 in autoimmune diseases will likely require long-term administration of IL-2. A recent preclinical study showed that chronic low-dose IL-2 treatment does not suppress beneficial effector immune responses, for example against infectious agents or in response to vaccination, at doses that prevent autoimmune diabetes.91 Another recent study found that low-dose IL-2 for GvHD prophylaxis after allogeneic HSCT reduced the frequency of infections and mediated the expansion of Tregs without diminishing its antiviral and anti-leukemic activity.92
Summary and Concluding Remarks
Autoimmune disease is chronic and debilitating, and there is an unmet need for safe and effective novel treatments. Autoimmunity is often characterized by dysregulation of CD4+ Tregs, which play a central role in immune tolerance and prevention of aberrant immune responses. Recent studies have improved our understanding of the role of IL-2 in restoring CD4+ Treg homeostasis and support a rationale for its use at low doses to induce Tregs and treat autoimmune diseases. Low doses of IL-2 in soluble form or coupled to anti-IL2 mAbs can also enhance Tregs, while avoiding stimulation of Teffs. A growing number of early phase clinical trials have provided initial safety and efficacy data in various autoimmune diseases but there remains a need for larger, long-term confirmatory studies. Nonetheless, based on extensive preclinical data and early clinical results, low-dose IL-2-based Treg therapy offers a promising avenue for the treatment of immune inflammation. Data from ongoing clinical trials are eagerly awaited.