Pancreatic cancer is estimated to become the second most common cause of cancer-related death by 2030, and most common by 2050 in the USA.1,2 Pancreatic cancer results in approximately 331,000 deaths annually worldwide, making it the seventh most common cause of cancer-related mortality.3 More than 90% of all pancreatic cancers are pancreatic ductal adenocarcinoma (PDAC).4 For all stages combined, PDAC has a very poor 5-year overall survival (OS) rate of 8%.5 Despite significant improvements in survival rates in many cancer types, PDAC death rate has increased in recent years.5 Recent advances in chemotherapy regimens have had a modest impact on median OS, which ranges between 8.5–11.1 months.6,7 Development of novel systemic therapies to overcome the resistance of PDAC is an urgent need. This article aims to provide an in-depth review of the literature around novel treatment strategies and ongoing research in pancreatic cancer management.
Overview of current systemic treatment approach to pancreatic ductal adenocarcinoma
Single-agent gemcitabine had been the established cytotoxic chemotherapy option based on clinical benefit defined as improvement in disease-related symptoms in patients with metastatic pancreatic cancer.8 The median OS with single-agent gemcitabine is 5.6 months. Several randomized trials combining different agents with gemcitabine have failed to provide any survival benefit.9–12 However, there have been some successful gemcitabine combinations; in a double-blind international phase III study, a statistically significant OS benefit was shown by gemcitabine plus erlotinib combination compared to gemcitabine plus placebo, 6.2 versus 5.9 months.13 Significant improvements in median OS have also been observed in two randomized trials – ACCORD 11 and MPACT (Metastatic Pancreatic Adenocarcinoma Clinical Trial) – both of which established the modern treatment regimens in the front-line setting.6,7 The ACCORD 11 trial compared FOLFIRINOX regimen (leucovorin, fluorouracil [5FU], irinotecan, and oxaliplatin) to single-agent gemcitabine. Median OS was 11.1 months versus 6.8 months, respectively. The multinational MPACT trial compared gemcitabine plus nab-paclitaxel combination to single-agent gemcitabine in newly diagnosed metastatic pancreatic cancer. Median OS was 8.5 months versus 6.7 months, favoring the combination arm. Both trials showed increased toxicities with the combination chemotherapy regimens as compared to gemcitabine. The current second-line treatment option was established by the NAPOLI-1 trial, which compared a nanoliposomal irinotecan plus 5FU combination to single-agent nanoliposomal irinotecan or 5FU in patients who were previously treated with gemcitabine-based chemotherapy.14 Median OS was 6.1 months in the combination arm, compared to 4.2 months with single-agent 5FU.15 More recently pembrolizumab was approved for microsatellite instability-high (MSI-H) tumors in the second-line setting based on data from 149 patients across five single-arm clinical trials.16
Other cytotoxic chemotherapy agents are being explored in clinical trials. The phase I/II NAPOX trial combined nanoliposomal irinotecan, 5FU/leucovorin, and oxaliplatin in the first-line setting, and reported promising anti-tumor activity – six partial responses in 24 patients in one cohort.17 Clinical trials with cytotoxic chemotherapy combinations such as gemcitabine, capecitabine, cisplatin, irinotecan and trifluridine/tipiracil, and nanliposomal irinotecan are underway (ClinicalTrials.gov identifier: NCT03535727).18
Despite modest improvements in OS with cytotoxic chemotherapy, the 5-year OS of PDAC is still 8% and there is an urgent need for novel, effective systemic treatment approaches.
What systemic therapies are there on the horizon beyond cytotoxic chemotherapy?
Molecular characterization and genomic profiling of PDAC has led to a better understanding of the disease and currently there are more than 150 actively accruing clinical trials with novel agents exploring new treatment options and breakthroughs. These novel clinical trials focus on immune system, targeted therapy, pancreatic stroma, and tumor microenvironment. Promising targets for therapy in pancreatic cancer include driver oncogenes, DNA repair pathway, stem cell, and metabolomic pathway.
Emerging therapeutic targets for pancreatic cancer
Immunotherapy
Immunotherapy with anti-programmed cell death protein 1 (PD-1), anti-programmed death-ligand 1 (PD-L1), and anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) antibody treatment has become standard of care in several malignancies; however, the outcomes in PDAC with single agent or dual immune checkpoint inhibitors have been disappointing.19,20 The profoundly immunosuppressive tumor microenvironment of PDAC, dense stroma, inhibitory cytokines, low effector T-cell, and low mutational tumor burden have been implicated in the failure of immunotherapy in pancreatic cancer.21,22 Additionally, the PDAC tumor microenvironment is infiltrated with immunosuppressor elements, such as myeloid-derived suppressor cells, tumor-associated macrophages (TAMs), and neutrophils. Patients with PDAC and high CD8 T-cell infiltration or neoantigen numbers have been shown to have longer survival.23 Presence of higher effector T cells and lower alternatively activated macrophages (M2) are also associated with longer survival.24,25 Other than in subgroups of patients with PDAC with MSI-H tumors, use of checkpoint inhibitors in PDAC is still investigational.
Microsatellite instability-high subgroup
Approximately 1% of patients with PDAC have MSI-H-positive disease.26 The immune checkpoint inhibitor pembrolizumab has shown a 53% objective response rate (ORR) across 12 solid tumor types, including pancreatic cancer.27 Pembrolizumab is currently approved for this indication in the second-line setting.27 Currently, a phase I study with anti-PD-L1 antibody LY3300054, alone or in combination with other agents, is enrolling patients with treatment refractory MSI-H solid tumors, including pancreatic cancer (ClinicalTrials.gov identifier: NCT02791334).
Immunotherapy and stromal targeting agents
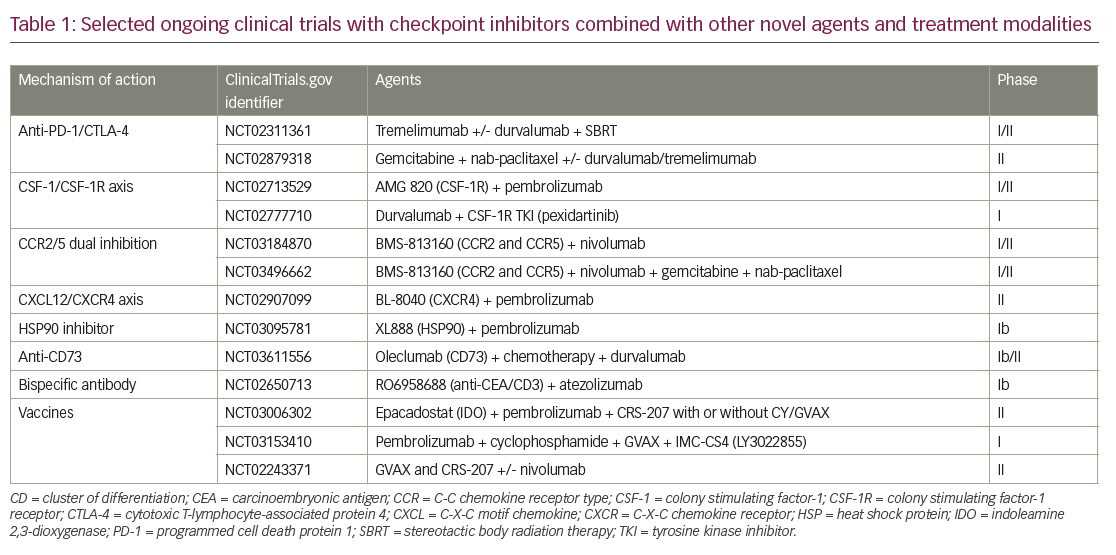
Two main approaches to enhance immune therapy effects in PDAC, are being evaluated. The first approach is aimed at enhancing the activity of the immune checkpoint blockade by adding agents that address the immune inhibitory environment. These include rational combination approaches with immunomodulatory chemotherapy agents, radiotherapy, and small molecular agents, focusing on the tumor microenvironment for a more immune-favorable state, improving antigen presentation, and decreasing regulatory T-cells (Table 1). In the second approach, investigators are evaluating immune therapy approaches that do not include checkpoint blockade.
Checkpoint inhibitor-based combination approaches
Chemotherapeutic agents such as gemcitabine and cyclophosphamide have been shown to decrease regulatory T cells,28 which prompted early-phase clinical trials in combination with checkpoint inhibitors (Table 1). A combination of Gemcitabine plus tremelimumab was studied in a phase Ib trial in metastatic PDAC, and two partial responses were reported.29
DNA damage is the main mechanism of cancer cell death by radiotherapy; however, more recently, the immunomodulatory effects of radiotherapy have been recognized.30 Radiation induces the release of proinflammatory mediators and damage-associated molecules such as high mobility group box 1 (HMGB1), heat shock proteins, ATP, and calreticulin;31 upregulates major histocompatibility complex class I expression, dendritic cell activation, immune checkpoint expression; and increases antigen presentation, cytotoxic T-cell recognition of irradiated cells, immunomodulatory cytokines, and inflammatory mediators.32,33 The synergistic effects of checkpoint-inhibitor and radiotherapy combination have been demonstrated in preclinical studies, and combination approaches are being explored in ongoing clinical trials (Table 1).34
TAMs have immunosuppressive and tumor-promoting features, and therefore have significant implications in carcinogenesis and progression of metastatic disease.35 TAMs and other myeloid cells secrete proangiogenic growth factors and immunosuppressive cytokines36 contributing to the immunosuppressive tumor microenvironment.35 TAMs can either have a proinflammatory (M1) or anti-inflammatory (M2) phenotype. An anti-inflammatory TAM M2 phenotype has immunosuppressive features and is associated with more aggressive tumors, increased angiogenesis, invasion, metastasis, and negative prognosis.37–39 The development of anti-inflammatory TAM M2 depends on growth factors such as colony stimulating factor-1 (CSF-1), also known as macrophage CSF. CSF-1 is activated by CSF-1 receptor (CSF-1R) and has crucial role in macrophage differentiation and survival.40 CSF-1R is expressed on myeloid-derived suppressor cells, neutrophils, and dendritic cells in the tumor microenvironment. CSF1-R mediated signals predominantly control the tumor promoting features of TAM; therefore, targeting tumor promoting CSF-1R is a viable anti-tumor strategy. Several clinical trials with CSF-R1 inhibitors in combination with checkpoint inhibitors, chemotherapy, and other immunotherapy agents are actively accruing patients (Table 1). Chemokines mediate immune-cell trafficking between bone marrow, peripheral tissues, inflammatory sites, and the tumor microenvironment.41 CCR2 and CCR5 are chemokine receptors that are expressed on myeloid cells in the tumor microenvironment and promote an immunosuppressive tumor microenvironment by recruiting inflammatory monocytes from bone marrow.42 CCR5 is also expressed on regulatory T cells and mediates their migration to the tumor microenvironment and promotes TAM M2 phenotype. Patients with PDAC with high C-C chemokine ligand (CCL)2 and low CD8 + T-cell infiltration have shorter survival compared to low CCL2 and high CD8 + T cells.43 Targeting the C-C chemokine receptor type (CCR)-2 axis with a CCR2 inhibitor has been shown to increase effector T cells, decrease regulatory T cells, and decrease tumor growth and metastasis in murine pancreatic cancer models.43 Anti-tumor effect was further increased when CCR inhibition was combined with gemcitabine in the same murine model. Based on this preclinical study, the oral CCR2 inhibitor PF-04136309 in combination with FOLFIRINOX or FOLFIRINOX alone was studied in a phase Ib trial in borderline resectable and locally advanced PDAC.43,44 A partial response rate of 48.5% was reported with the combination therapy and treatment was tolerable.44 Investigation into a CCR2/CCR5 dual inhibitor in combination with gemcitabine, nab-paclitaxel, and nivolumab in borderline resecable or locally advanced PDAC is ongoing (ClinicalTrials.gov identifier: NCT03496662). A trial with CCR2/CCR5 dual antagonist in combination with chemotherapy or nivolumab is also ongoing in metastatic pancreatic and colorectal cancer.42
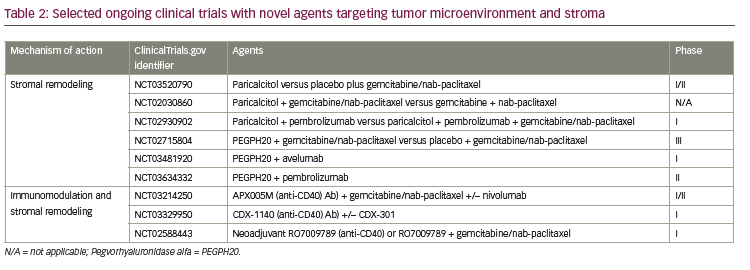
Tumor microenvironment and stroma
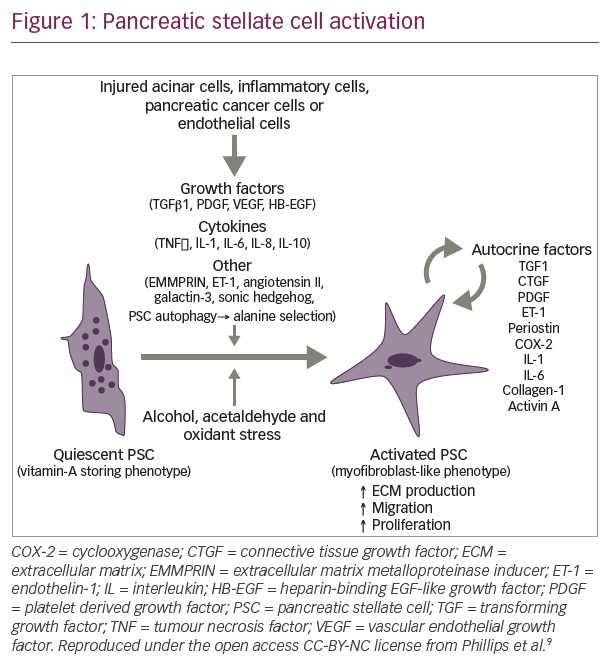
Dense stromal desmoplastic reaction is one of the hallmarks of pancreatic cancer tumor microenvironment.45 The fibrous tissue which constitutes the desmoplastic reaction is produced by pancreatic satellite cells. Pancreatic satellite cells also produce extracellular matrix proteins, cytokines, and vascular endothelial cells.21,46 The stromal desmoplasia is evident in both primary pancreatic tumors and metastatic tumor sites and creates a more hypoxic tumor microenvironment.47 In PDAC, the stroma is highly fibrotic, which creates a hypoxic tumor microenvironment and is implicated in tumor progression, metastasis, and treatment resistance; although it can have tumor restraining effects.21,48 Novel strategies targeting the stroma and tumor microenvironment, involving hedgehog pathway and Bruton’s kinase, have resulted in disappointing outcomes.49,50 Current strategies are focusing on targeting hyaluronic acid (HA), vitamin D receptor, and CD40 antibody (see Table 2).
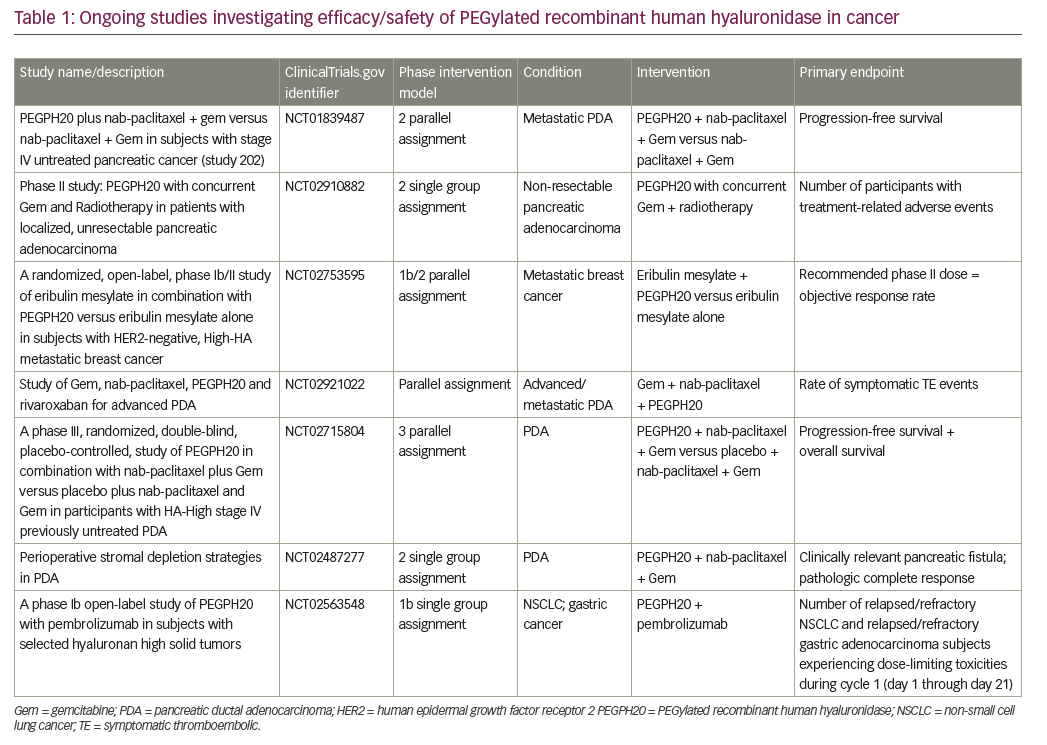
HA is abundant in pancreatic stoma; it increases interstitial pressure, impacts oncogenesis, and is involved in cellular signaling pathway by binding to cell surface receptors, such as CD44.51–53 Pegvorhyaluronidase alfa (PEGPH20), a pegylated version of recombinant human hyaluronidase, has been shown to decrease HA enzymatically, and has reestablished and improved vascular permeability and improved intratumoral drug delivery in a preclinical study.54 PEGPH20 in combination with gemcitabine also resulted in higher survival and reduction in tumor size compared to gemcitabine alone in the same mouse model.54 The phase II HALO 202 trial compared PEGPH20 plus nab-paclitaxel/gemcitabine versus nab paclitaxel/gemcitabine regimen in untreated metastatic PDAC and reported improved progression-free survival (PFS), 6 months versus 5.3 months, respectively.55 When patients with HA-high tumors were analyzed, both the PFS and ORR was higher in the PEGPH20 plus nab-paclitaxel/gemcitabine arm, 9.2 months and 45% versus 5.2 months and 31%, respectively. Due to a higher rate of thromboembolic events in the PEGPH20 plus nab-paclitaxel/gemcitabine arm, this study was amended after the first stage, and enoxaparin prophylaxis was implemented in both arms. A clinical trial with PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in HA-high PDAC is ongoing (ClinicalTrials.gov identifier: NCT02715804). Contrary to favorable outcomes with the GA combination, a phase I/II trial with PEGPH20 plus mFOLFIRINOX versus mFOLFIRINOX in untreated PDAC population was recently closed to accrual due to detrimental effect of OS.56
CD40 belongs to the tumor necrosis factor superfamily and is expressed by antigen-presenting cells as well as tumor cells.57 CD40 activation stimulates antigen presentation by the antigen-presenting cells, triggers proinflammatory cytokine release, enhances T-cell activation, and induces stromal depletion in both preclinical pancreatic cancer models and humans.57,58 The synergistic effects of CD40 agonist and cytotoxic chemotherapy combination has been shown in preclinical studies when CD40 was administered following gemcitabine.59 In another preclinical study with a KPC mouse model, CD40 activation redirected tumor infiltrating monocytes and TAMs and induced degradation of fibrosis, hence increased efficacy of chemotherapy.60 Interestingly, the sensitivity to chemotherapeutic agent was enhanced even when administered just days after CD40 antibody, indicating rapid degradation of fibrosis in the tumor model. The immunomodulatory effects of CD40 in the tumor microenvironment and antifibrotic properties in tumor stroma prompted further exploration of anti-CD40 monoclonal antibodies in clinical trials (Table 2). In a phase I study of 22 patients with advanced PDAC, the combination of agonistic CD40 monoclonal antibody with gemcitabine was well tolerated and achieved 19% ORR.61
PDAC stroma expresses vitamin D receptor; in preclinical study with a KPC tumor model, vitamin D analogue, calcipotriol, was shown to induce stromal remodeling, increase intratumoral vasculature, improve intratumoral drug delivery, and increase anti-tumor response when combined with gemcitabine.62 A phase II study combining with gemcitabine, cisplatin, nab-paclitaxel, paricalcitol, and nivolumab is currently enrolling; preliminary results from 10 patients in the initial phase of the study have revealed 80% ORR.63 A first-line placebo-controlled phase II clinical trial with the vitamin D agonist, paricalcitol, plus gemcitabine/nab-paclitaxel is currently accruing (ClinicalTrial.gov identifier: NCT03520790). Pilot studies are being conducted in resectable pancreatic cancer with vitamin D analogs and chemotherapeutic agents (ClinicalTrial.gov identifier: NCT02030860) and with checkpoint inhibitors (ClinicalTrial.gov identifier: NCT02930902).
Targeting DNA damage response and other pathways
Pancreatic adenocarcinoma has a complex genomic landscape and further molecular characterization has led identification of unique subgroups with different mutational features which may benefit from molecularly targeted therapy. Waddel et al. performed whole-genome sequencing of 100 patients with PDAC and demonstrated that chromosomal structure variation is a significant mechanism of DNA damage in PDAC.64 As such, PDAC has been classified into four different subtypes: stable subtype, locally rearranged subtype, scattered subtype, and unstable subtype. Scattered subtype has <200 structural abnormalities whereas unstable type has >200 structural abnormalities and mutations in DNA repair pathways such as BRCA1/2, PALB2, and ATM, which are more sensitive to platinum-containing regimens. A recent comprehensive genomic analysis of 456 pancreatic cancer cases revealed commonly mutated genes that aggregate into 10 cellular pathways.65 Based on gene expression, four different subtypes have been identified including squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine. The gene expression features of these subtypes were correlated with histopathological characteristics and survival.
KRAS mutation is seen in 95% of PDACs and TP53, CDKN2A, SMAD4/DPC4, MLLSM, and RBM10 are other well-known mutations; however, the frequency of potentially actionable mutations with effective targeted therapies is much lower.66,67 Certain mutations such as NTRK gene fusions are highly actionable with very effective targeted therapy but NTRK gene fusions are detected in less than 1% of PDACs.68,69 Prominent investigational targeted therapy approaches include DNA repair pathways, cancer stem cells, metabolic pathways, and asparagine depletion.
DNA repair
Molecular subtyping of pancreatic cancer may have therapeutic implications as different subtypes may respond to therapy differently.70 Disrupted DNA damage response (DDR) is one of the hallmarks of pancreatic cancer development and is seen in up to 24% of PDACs,66 which also opens avenues for cancer treatment by utilizing synthetic lethality by targeting complementary DNA repair mechanisms by platinum agents and PARP inhibitors.71 BRCA1/2, PALB2, and ATM genes are key DNA maintenance genes in pancreatic cancer development.72 The majority of unstable type tumors have shown high BRCA mutational signature and/or unstable genome.64 BRCA-associated PDAC has superior survival with platinum-containing regimens and responds better to PARP inhibitors.73 Significant clinical activity with an ORR of 77.8% with PARP the inhibitor veliparib in combination with gemcitabine and cisplatin was reported in a phase I clinical trial in BRCA-mutated PDAC.74 Currently, a randomized phase II trial with and without veliparib combined with gemcitabine and cisplatin is enrolling patients (ClinicalTrials.gov identifier: NCT01585805) along with several other trials with PARP inhibitors (Table 3). The POLO trial randomized patients with BRCA-mutated metastatic PDAC to olaparib versus placebo (3:2 ratio) as a maintenance therapy after disease stability on a platinum containing regimen.75 The study has achieved its primary endpoint of improvement in PFS, which establishes an evidence-based approach for maintenance therapy in platinum sensitive BRCA-mutated PDAC. DDR mutations such as ATM, ATR, ATRX, BAP1, BARD1, BRIP1, CHEK 1/2, RAD50/51B, FANCA/C/D2/E/F/G/L beyond BRCA 1/2, and PALB2 are associated with improved OS when treated with platinum-containing regimens in advanced pancreatic cancer.76

Cancer stem cells
Cancer stem cells are highly tumorigenic, they have the ability to self-renew, can differentiate into heterogenous nontumorigenic cancer cell types comprising the tumor, are able to form spheres in stem cell media, and are resistant to traditional chemotherapeutic agents.77–80 A preclinical study with cancer stemness inhibitor BBI608, napabucasin, in a PaCa-2 pancreatic xenograft model, inhibited tumor growth, decreased stemness-high cancer cells in vivo, and down-regulated cancer stem cell proliferation and STAT3-driven self-renewal genes as compared to gemcitabine and carboplatin.78 In a phase Ib extension study with napabucasin and gemcitabine/nab-paclitaxel combination, no dose limited toxicity was encountered; in 29 evaluable patients, a 93% disease control rate and 79.3% ORR was reported in previously untreated PDAC.81 In a phase Ib/II study with the same combination, in 55 evaluable patients, a 93% disease control rate and 55% ORR was reported.82 Based on these encouraging results, a phase III trial with nababucasin plus gemcitabine/nab-paclitaxel is currently ongoing.83
Metabolic pathways
The mitochondrial metabolism inhibitor CPI-613 targets mitochondrial energy metabolism in cancer cells and induces apoptosis and autophagy.84 In a preclinical pancreatic cancer xenograft tumor model, significant anti-tumor activity was observed.84 A phase I trial with CPI-613 combined with mFOLFIRINOX reported 61% ORR, with hyperglycemia, hypokalemia, and peripheral neuropathy being the most common grade 3–4 nonhematological adverse events.85 CPI-613 is being explored in a phase III trial in combination with mFOLFIRINOX (ClinicalTrials.gov identifier: NCT03504423) and phase I trial with in combination with gemcitabine/nab-paclitaxel (ClinicalTrials.gov identifier: NCT03435289).
Erythrocyte-encapsulated L-asparaginase (eryaspase) has emerged as a potential therapeutic option for PDAC. Asparagine has an essential role in protein and nucleotide synthesis, it also regulates apoptosis, cell proliferation, and is essential for survival of pancreatic cancer cells.86–88 Asparagine synthase (ASNS) is induced by ATF4 and synthesizes asparagine from aspartate. Intracellular depletion of asparagine was shown to induce apoptosis.89 L-asparaginase depletes extracellular asparagine and it has anti-cancer effect on cancer cells with no or low ASNS expression.87 Interestingly, in lung cancer preclinical models, KRAS was identified as an important regulator in nutrient stress response in the cell, and through ATF4 regulation it was shown to alter asparagine biosynthesis, which is relevant in protein biosynthesis and apoptosis suppression.90 In the same preclinical study, ASNS was noted to be a key regulator of tumor cell proliferation. Disruption of the KRAS–ATF–ASNS pathway by inhibition of AKT sensitized tumor cells to L-asparaginase and was proposed as a potential treatment strategy in lung cancer. Eryaspase is encapsulated inside a donor-derived red blood cell, and anti-tumor activity by plasma asparagine depletion has been reported in a preclinical PDAC study.91 In a phase IIB study with chemotherapy +/- eryaspase, significant improvements in PFS and OS (26.1 for chemotherapy plus eryaspase versus 19.0 weeks for chemotherapy alone, p=0.03) were shown regardless of ASNS expression levels.92 An ongoing randomized phase III trial is exploring gemcitabine/nab-paclitaxel or an irinotecan-based regimen with or without eryaspase in second-line PDAC (ClinicalTrials.gov identifier: NCT03665441).
Conclusion
Clinical management of metastatic PDAC still depends on cyctotoxic chemotherapy. Although prevalence of MSI-H and NTRK gene fusions is low in PDAC, FDA-approved molecularly targeted therapy options are available for this subgroup of patients. Several promising therapies are in development based on strong scientific rationale including combination therapies with immune checkpoint inhibitors, stroma modifying agents, targeting of TAMs, DNA repair pathways, and cancer stem cells.