Pancreatic ductal adenocarcinoma (PDA) is predicted to beat colon cancer in terms of total cancer-related deaths in the USA by this year (2020).1 Given this projected burden of disease, efforts in understanding and developing new treatments for PDA are paramount. In this review, we will summarize the molecular characteristics and mutations thought to be involved with PDA oncogenesis, discuss current PDA outcomes, list recent clinical progress, and comment on how we can improve outcomes in PDA.
Molecular characterization of pancreatic cancer
Pancreatic cancer usually begins as one of three precursor lesions: pancreatic intraepithelial neoplasia (PanIN), intraductal papillary mucinous neoplasms (IPMNs), or mucinous cystic neoplasms. For brevity, in this review, since the majority of pancreatic cancer stems from PanINs, we will focus on epithelial neoplasias. IPMNs account for 1–3% of exocrine neoplasms and up to 50% of cystic pancreatic neoplasms,2 while mucinous cystic neoplasms can also progress to adenocarcinoma, but at lesser frequency.3 Traditionally, PDA oncogenesis is thought to be initiated by a pre-existing PanIN, which undergoes one of four classic mutations known to be involved in pancreatic cancer. These four mutations are KRAS oncogene activation, TP53, p16/CDKN2A, or SMAD4 tumor suppressor inactivation.4 A PanIN combined with (most frequently) a KRAS mutation is the hypothesized driver mutation, which leads to further mutations, causing genomic instability and promoting pancreatic carcinogenesis.
Today, we have evidence for a wider variety of somatic mutations likely involved in pancreatic cancer, such as: KMT2C, TGFBR2, ARID1A, EPC1, ARID2, SF3B1, ATM and RNF43.5 There are also germline mutations associated with hereditary syndromes, including MLH1 and MSH2 (associated with Lynch syndrome), BRCA2,6 PALB2 (involved with the Fanconi anemia pathway),6 STK11 (associated with Peutz–Jeghers syndrome),7 and ATM (associated with ataxia–telangiectasia).5 These have also been implicated in the oncogenesis of some hereditary forms of PDA.
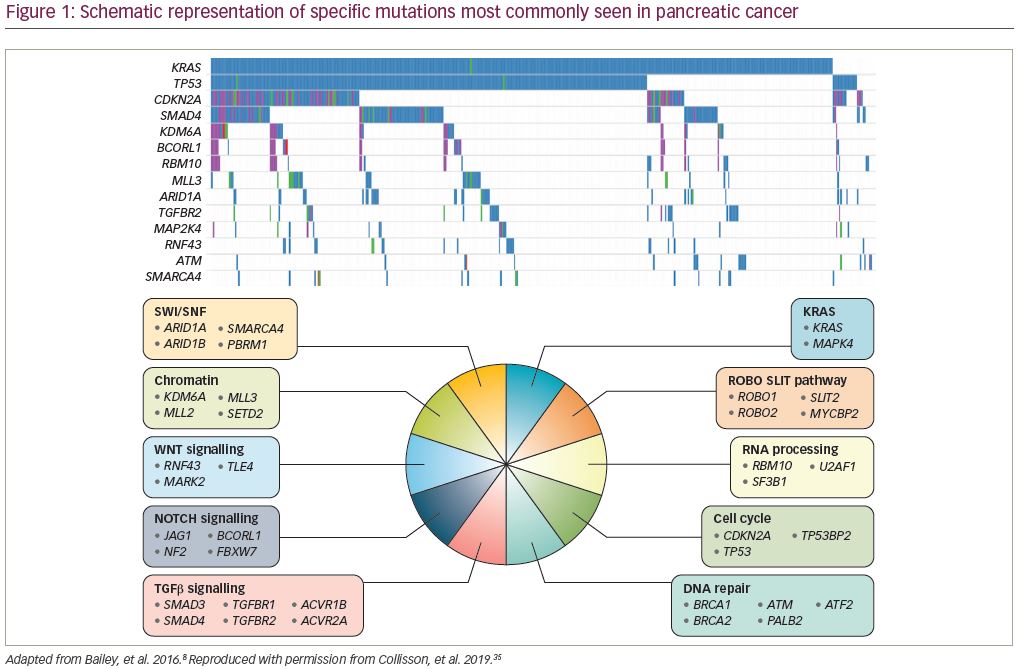
From a larger-scale perspective, these mutations are part of a number of dysregulated cellular pathways that have been implicated in pancreatic cancer. Bailey et al. described these molecular subtypes by genomic analysis of 456 tumors. They found that the pathways involved centered around apoptosis, G1/S phase transition, hedgehog signaling, TGF (transforming growth factor) signaling, Wnt/Notch (wingless-related integration site/notch) signaling, ROBO/SLIT (Roundabout receptor/Slit protein) signaling, SWI-SNF (SWItch/Sucrose Non-Fermentable) ATP-dependent chromatin remodeling complexes, chromatin modification, DNA repair, and RNA processing.8 Tumor mutational pathways have also been organized by subtype by Bailey et al. via gene expression analysis, as well as by potential mutational targets (Figure 1). The four subtypes are: (1) squamous, (2) pancreatic progenitor, (3) immunogenic, and (4) aberrantly differentiated endocrine exocrine, which correlate with histopathological characteristics.8 Pending further research, these genomic and transcriptomic subtypes may be able to help guide future therapy efforts.
Another driver of pancreatic cancer tumorigenesis is large-scale “catastrophic” mitotic events,9 which lead to rapid tumorigenesis. These large-scale events may be responsible for the rapid speed of tumor formation, ultimately meaning that we have less time for tumor detection. This would also explain why PDA progresses and metastasizes quickly once it has transformed to a malignant tumor.9 Large genomic rearrangement patterns include: polyploidy/aneuploidy, deletion, inversion, tandem duplication/end-to-end chromosome fusions with telomere erosions, amplicon, foldback inversions, and chromothripsis (or breakage of thousands of chromosomal segments follow by rejoining in an incorrect orientation).9–11 These are important mechanisms in understanding pancreatic tumor oncogenesis, because a certain subset of patients may have this “punctuated equilibrium” occur, instead of a gradual and stepwise mutation order,9,12 which typically describes other types of cancers.
Current treatment, outcomes, and challenges
First-line treatment for resectable pancreatic cancer is surgery with adjuvant chemotherapy. For non-resectable cases, candidates for chemotherapy are treated based on performance status with FOLFIRINOX (leucovorin calcium [folinic acid], fluorouracil, irinotecan hydrochloride, oxaliplatin), or, if the patient cannot tolerate FOLFIRINOX, varying doses of gemcitabine with/without nab-paclitaxel. Patient fitness is also relevant to the aforementioned standard of care chemotherapy. FOLFIRINOX, more so than gemcitabine, has a harsh grade 3 or 4 adverse event profile, including neutropenia, fatigue, vomiting, diarrhea, neuropathy, and transaminasemia. This is due, in particular, to the fluorouracil component and less so to oxaliplatin (but better tolerated than cisplatin). Given the difficult side effects, the regimen requires patients to have a higher functional status prior to starting therapy, in order to withstand treatment.
Adjuvant chemotherapy was established as standard of care after the CONKO-001 study showed significantly increased disease-free survival (DFS) and overall survival (OS) in patients treated with 6 months of gemcitabine (versus observation only) after complete resection of their tumor.13 Median DFS was 13.4 versus 6.9 months, (p<0.001), and median OS was 22.8 versus 20.2 months (p=0.005) in the gemcitabine versus observation group, respectively. Furthermore, estimated survival at 3 and 5 years was 36.5% and 21.0% for gemcitabine-treated patients versus 19.5% and 9.0% for observation post-resection.13 For a complete list of clinical trials discussed in this review, see Table 1.
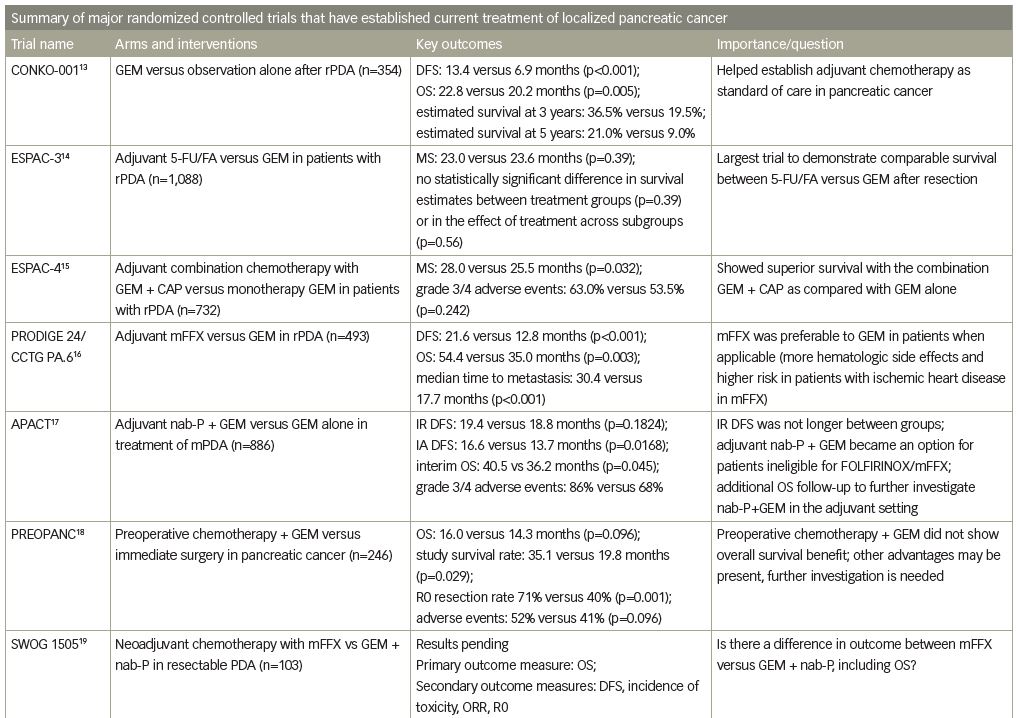
After adjuvant gemcitabine was established as the gold standard, the ESPAC-3 trial broadened chemotherapy options by showing comparable survival between 5-fluorouracil and leucovorin versus gemcitabine after resection.14 ESPAC-4 further showed superior survival with the combination of gemcitabine plus capecitabine, an oral fluoropyrimidine, compared to gemcitabine alone.15 Most recently, the PRODIGE 24/CCTG PA.6 study demonstrated an increased DFS in patients treated with modified FOLFIRINOX (mFFX) compared with gemcitabine (21.6 versus 12.8 months) and, more strikingly, OS was significantly improved (54.4 versus 35.0 months).16
Surprisingly, while the combination of gemcitabine and nab-paclitaxel is routinely used in the metastatic setting, it does not seem to have the same benefit in the adjuvant space. The APACT trial found no DFS difference between adjuvant nab-paclitaxel/gemcitabine versus gemcitabine alone.17 Of note, the primary endpoint for this study was an independently assessed DFS, a method which was not used in the prior adjuvant trials, and the OS data are still maturing.17
With respect to resectable pancreatic cancer, the pendulum is swinging more toward neoadjuvant chemotherapy followed by surgery. This may be due, in part, to the fact that pancreatic cancer is really a systemic disease, and the field of surgical oncology has started to further incorporate this thought into practice with neoadjuvant therapy. For example, in PREOPANC, patients received either adjuvant chemotherapy with gemcitabine or immediate surgery. There was improved survival (85.2 versus 19.8 months), and although median survival did not significantly differ (16.0 versus 14.3 months, respectively), the R0 (negative margin) resection rate (71% versus 40%), DFS, and distant metastasis-free interval were all significantly improved in the neoadjuvant chemotherapy group.18 The ongoing SWOG 1505 study compares perioperative (12 weeks pre-op, 12 weeks post-op) chemotherapy with either mFFX or gemcitabine plus nab-paclitaxel.19 The study’s primary outcome of interest is 2-year OS.19 Given the efficacy of both regimens in the metastatic setting, this study will hopefully guide us in the chemotherapy combination of choice for localized resectable pancreatic cancer.
Improving outcomes
Screening
Potential signs of PDA on clinical examination are limited due to the anatomical location of the pancreas. In addition, symptoms of PDA are nonspecific, such as asthenia, weight loss, anorexia, abdominal pain, or jaundice. Risk factors for PDA are broad, such as cigarette smoking; 11–32% of pancreatic cancer deaths are attributed to tobacco smoking.20 High body mass, lack of physical activity, a “Western” dietary pattern, and heavy alcohol use are also associated with high risk of pancreatic cancer.21–23 In addition, based on 3D models, pancreatic cancer appears to metastasize early, often before it is even diagnosed.24 This may be based on simple pathophysiology, as early-stage pancreatic cancer invades its surrounding vasculature, draining into the hepatic circulation, and thus results in early metastatic spread.11
There is no standard diagnostic tool or method for early detection of pancreatic cancer, and the benefit of early detection may be questionable. A good screening program needs not only a sensitive-then-specific method, but also, if we do catch PDA early, then early detection needs to make a difference in treatment/survival compared with later detection of PDA. We currently do not have well-founded evidence for this. With adjuvant FOLFIRINOX after surgery, we may make a difference in mortality by early detection, but this will take further research.
At this time, the only US Food and Drug Administration (FDA)-approved tumor marker for pancreatic cancer diagnostics is CA 19-9,25 but it is neither sensitive nor specific. The Cancer of the Pancreas Screening (CAPS) studies were designed to review the best prevention, screening, and surveillance methods for individuals at high risk for pancreatic cancer, and describe them in a general guideline format. According to the CAPS guidelines, candidates for screening would be: first-degree relatives of patients with pancreatic cancer, or any family member with at least two known first-degree relatives with pancreatic cancer; people with Peutz–Jeghers syndrome; and p16, BRCA2 and hereditary non-polyposis colorectal cancer (HNPCC) mutation carriers with at least one affected first-degree relative.26 Additionally, screening methods would ideally be able to detect T1N0M0 tumors as well as high-grade dysplastic precursor lesions (PanIN and IPMNs) at risk for malignant transformation.26 However, there is no consensus from the CAPS studies as to what age group to start and stop pancreatic cancer screening, and which imaging modality is best to use for screening, though generally, endoscopic ultrasonography and/or magnetic resonance imaging (MRI) cholangiopancreatography is used initially.26
Liquid biopsy and monitoring
The diagnosis of almost any cancer is dependent on biopsy confirmation. For pancreatic cancer, after high-quality computed tomography (CT) or MRI, endoscopy for tissue and molecular testing, and laparoscopy to rule out occult peritoneal disease, invasive endoscopic fine needle aspiration can confirm the histopathology. Research on new diagnostic methods include the use of circulating tumor cells (CTCs).27 This is promising for pancreatic cancer, as PDA CTCs may be present even before the tumor is detectable by current methods.27 CTC liquid biopsy could eventually become a minimally invasive surrogate for an invasive biopsy via endoscopy.27 One notable advantage of a liquid biopsy is the ability to follow tumor changes in “real time”—by giving an objective update on the heterogeneity of an individual’s tumor at different time points (for example, pre-treatment, post-treatment etc).27 Other advantages of using CTCs in pancreatic cancer include the fact that CTCs have been detected in all stages of PDA,28 CTCs are present in precancerous lesions and before a tumor can be detected by imaging,29 and CTCs are in fact at significantly lower levels in healthy individuals compared with people with pancreatic lesions.30,31
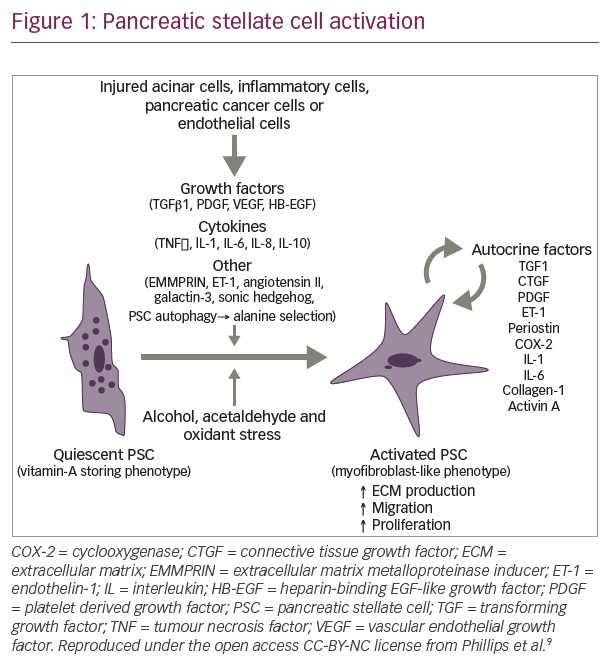
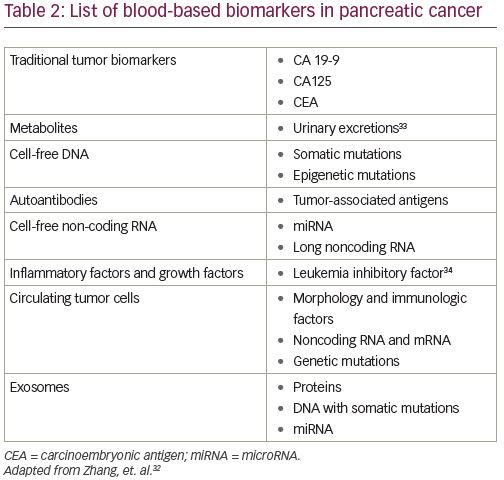
Other blood-based biomarkers being evaluated for early detection include the following categories: metabolites (such as cell-free DNA, autoantibodies, cell-free non-coding RNA, inflammatory factors, growth factors, exosomes,32 and other urine biomarkers33). Another promising biomarker for diagnosis and treatment response is leukemia inhibitory factor (LIF). LIF is a key paracrine factor released by pancreatic stellate cells, which have a close, reciprocal relationship with connecting pancreatic tumor cells.34 In human pancreatic cancer, aberrant production of LIF occurs only in pathological conditions, correlates with PDA pathogenesis, and LIF levels also correlate with a tumor’s response to therapy.34 Pharmacological blockade and genetic deletion of the LIFR gene has been shown to slow tumor progression and improve the efficacy of chemotherapy in mouse models with pancreatic cancer via modulation of cancer-cell differentiation and epithelial-mesenchymal transition status.34 A more complete list of blood-based biomarkers is listed in Table 2.
Current treatment research
Advances in PDA are being made on all fronts, in localized and metastatic disease. A myriad of molecular targets are under active investigation,35 utilizing a broad spectrum of tactical approaches. In resectable PDA, the foremost treatment is of course, resection, and we applaud the work of surgical oncology. For the sake of brevity, we will mostly focus on research from a medical oncologic perspective, mostly involving human subjects only.
Locally advanced pancreatic cancer studies
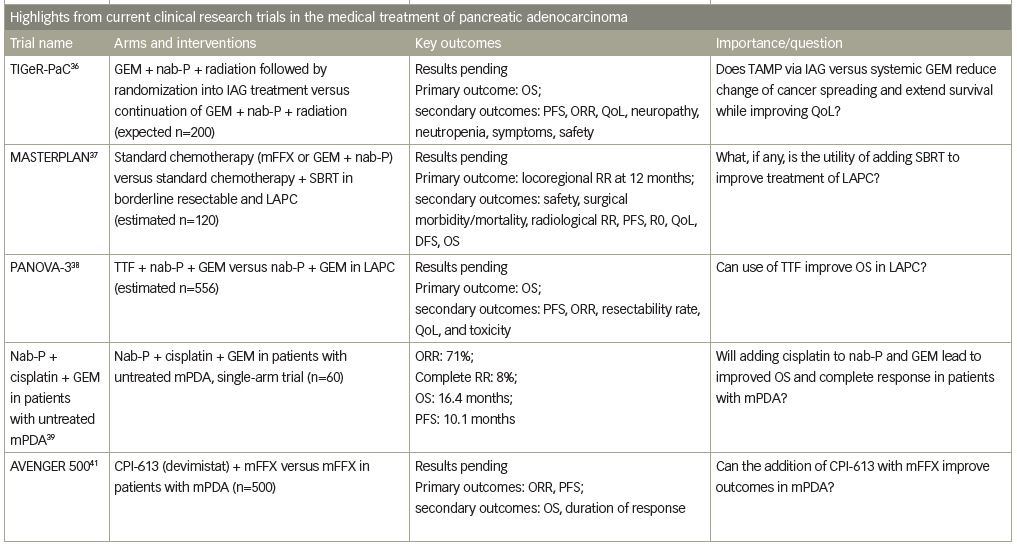
Locally advanced pancreatic cancer treatment is usually limited to systemic therapy with FOLFIRINOX or gemcitabine with/without nab-paclitaxel, as previously mentioned. Today, clinical trials are using novel therapies, such as innovative techniques to better target tumor cells. For example, a phase III clinical trial, TIGeR-PaC, is using intra-arterial delivery of gemcitabine (IAG) for local disease. The trial results showed that in previously radiated tumors, the local microvasculature had increased efficacy of IAG.36 Patients received induction therapy with intravenous gemcitabine + nab-paclitaxel + radiation therapy, and those without disease progression were divided into IAG treatment group versus continuation of the gemcitabine + nab-paclitaxel + radiation.36 The primary outcome measure is OS, and results are pending.
Several studies are evaluating the utility of adding stereotactic body radiation therapy (SBRT) and other local methods to improve treatment of locally advanced pancreatic cancer. For example, the MASTERPLAN study will compare standard (systemic) chemotherapy (mFFX or gemcitabine + nab-paclitaxel) versus standard chemotherapy + SBRT in both borderline resectable and locally advanced (non-resectable) tumors. The primary endpoint is locoregional response rate at 12 months.37
Tumor treating fields (TTF) is FDA-approved for the treatment of glioblastoma multiforme.38 TTF uses alternating electric fields from a transducer, targeted to tumor location, and disrupts cell division.38 The PANOVA-3 phase III clinical trial is randomizing patients with locally advanced pancreatic cancer to TTF + nab-paclitaxel + gemcitabine versus nab-paclitaxel + gemcitabine alone. Similar to glioblastoma multiforme, the primary endpoint is OS, but secondary endpoints include progression-free survival (PFS), objective response rate, resectability rate, quality of life, and toxicity.38
Metastatic pancreatic ductal adenocarcinoma studies
Building on the gemcitabine and nab-paclitaxel backbone, a recent study on nab-paclitaxel + cisplatin + gemcitabine in patients with untreated metastatic PDA, demonstrated the efficacy of adding cisplatin for patients with previously untreated metastatic PDA. This single-arm phase Ib/II study demonstrated an impressive overall response rate of 71%, and the disease control rate was 88%.39
Drug targets, via antibodies, are also being studied in the metastatic setting. A phase Ib study using a CD40 agonist antibody showed promising results in combination with nab-paclitaxel + gemcitabine with or without nivolumab. Post-baseline tumor results showed low CD8 T cell and high macrophage infiltrate in two of six patients, and circulating KRAS DNA decreased after therapy in some patients, revealing remodeling of the myeloid response to treatment.40 Another novel drug under investigation is devimistat (CPI-613®, Rafael Pharmaceuticals, Cranbury, NJ, USA), a lipoate analog that inhibits pyruvate dehydrogenase and α-ketoglutarate dehydrogenase enzymes, thus inhibiting the tricarboxcylic acid cycle and mitochondrial respiration.40 Early studies with devimistat in combination with chemotherapy showed an objective response rate of 61% and a 17% complete response rate.40 An ongoing phase III study, titled AVENGER 500, is looking at outcomes of CPI-613 + mFFX versus mFFX alone in patients with newly diagnosed metastatic PDA.41
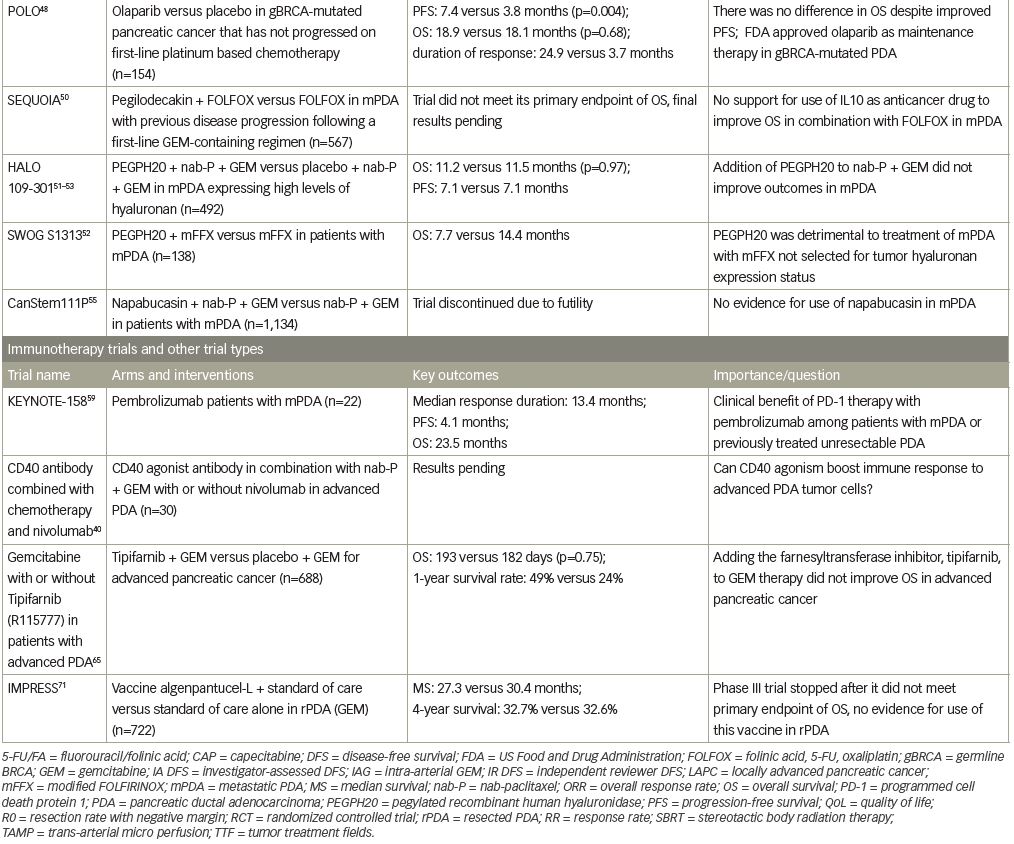
BRCA1 and 2 are autosomal-dominant tumor-suppressor genes with incomplete penetrance that are associated with familial pancreatic cancer.42 About 2% of patients with PDA are positive for the BRCA2 mutation, and ≤1% are positive for the BRCA1 mutation.43–45 Among these, patients with germline BRCA1 or BRCA2 mutations may be candidates for the use of olaparib, a poly ADP ribose polymerase (PARP) inhibitor. PARP enzymes help repair single-stranded DNA breaks (SSB), but are also found to be trapped in DNA at sites of SSBs (likely at unligated Okazaki fragments),46 which explains the lethal effect of PARP inhibitors.47 Recently, the POLO phase III trial randomized patients with metastatic PDA with germline BRCA mutations to receive the PARP inhibitor olaparib versus placebo as maintenance therapy after response or stability on platinum-based chemotherapy. Olaparib treatment delayed progression of disease compared with placebo (median PFS 7.4 versus 3.8 months, respectively, hazard ratio 0.53; p=0.004).48 Ultimately, results showed no difference in OS between the groups; which, unfortunately, meant improved PFS without improved OS. It is impressive to note that the response to olaparib versus placebo was 23% versus 12%, with duration of response to olaparib being 24.9 versus 3.7 months versus placebo.48 Based on these data, olaparib has been approved by the FDA as maintenance therapy after response or stable disease on platinum chemotherapy in BRCA-mutated PDA.48 Newly published phase II studies have shown no improvement in relative risk with concurrent veliparib (another PARP inhibitor), and increased grade 3/4 hematologic toxicities, but did support cisplatin and gemcitabine as a standard approach to first-line treatment in BRCA-positive PDA.49
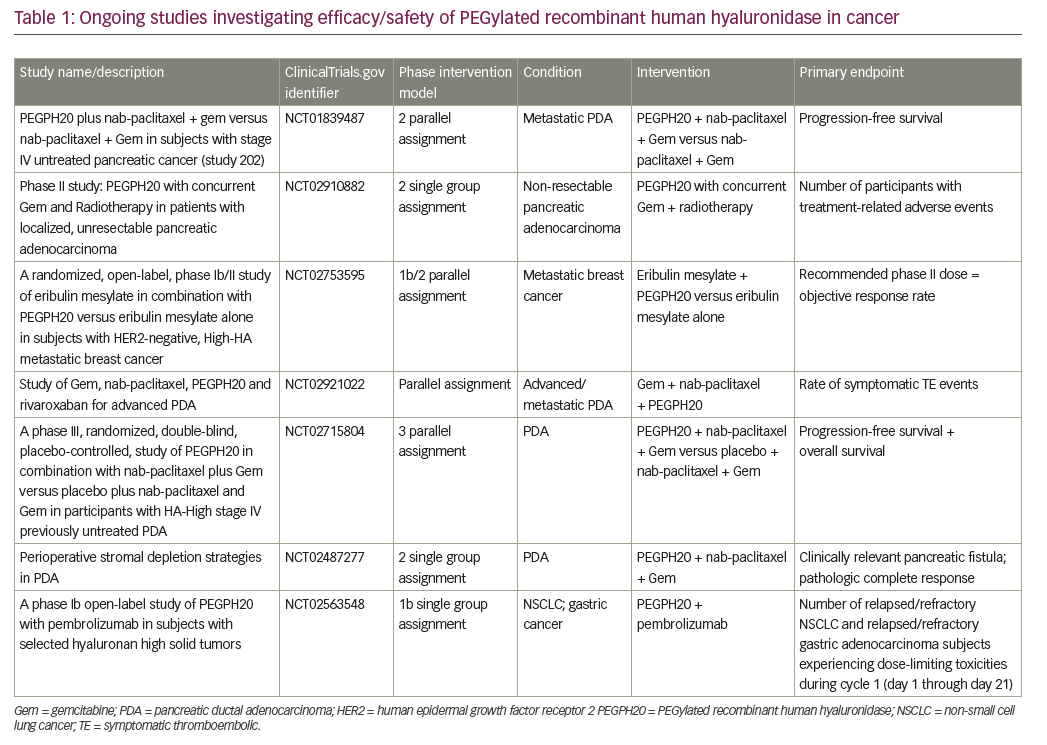
Unfortunately, a number of recent pivotal phase III studies have also reported negative results. The SEQUOIA trial evaluated pegilodecakin (pegylated interleukin [IL]-10) + FOLFOX (folinic acid, 5-FU, oxaliplatin) compared with FOLFOX alone in metastatic pancreatic cancer with previous disease progression following a first-line gemcitabine-containing regimen. The trial did not meet its primary endpoint of OS.50 Other unsuccessful efforts include targeting the pancreatic tumor microenvironment via hyaluronic acid inhibition. Hyaluronic acid is hydrophilic and, when present in high levels inside a tumor, increases interstitial pressure and thus blocks access of anticancer drugs to the tumor via reduced perfusion.51 Pegylated recombinant human hyaluronidase (PEGPH20) would thus inhibit hyaluronic acid from accumulating in the tumor microenvironment. The SWOG S1313 study showed worse outcomes, including reduced OS with the addition of PEGPH20 to FOLFIRINOX, although these patients were not selected based on hyaluronan expression status.52 Similarly, in the HALO 109-301 study, the combination of PEGPH20 + nab-paclitaxel + gemcitabine showed no improvement in OS, PFS, or duration of response, as compared with placebo + nab-paclitaxel + gemcitabine in patients with pancreatic cancer expressing high levels of hyaluronan.53 Finally, Napabucasin (BBI-608) is thought to inhibit the elusive cancer stem cell via the signal transducer and activator of transcription 3 (STAT3) pathway.54 The CanStem111P trial looked at the efficacy and safety of napabucasin in metastatic PDA.55 This phase III trial compared napabucasin + nab-paclitaxel + gemcitabine versus nab-paclitaxel + gemcitabine alone, but unfortunately, the trial was discontinued due to futility.55
Immunotherapy possibilities
In the last several years, immunotherapy has come into the spotlight as a game-changing approach to cancer treatment. While a number of drugs have been FDA approved in other malignancies, such as melanoma and non-small-cell lung cancer, at this time, there are no formally approved immunotherapy drugs for PDA, as studies with single-agent checkpoint inhibitors have been disappointing.56 This is thought to be explained by inherently low immunogenicity of PDA and the immunosuppressive tumor microenvironment of pancreatic tumors.57
One exception to the use of immunotherapy alone in PDA is in high microsatellite instability (MSI-H) cases. MSI-H is defined as microsatellite testing that shows mutations in ≥30% microsatellites.58 Unfortunately, MSI-H is a rarity in PDA and seen mostly in germline mutations or epigenetic inactivation of the MLH1 gene.58 This is important to mention because pembrolizumab, a highly selective anti-PD-1 monoclonal antibody, which reverses T-cell suppression and induces antitumor response,59 was evaluated for use in patients with MSI high PDA (which accounted for almost 10% of participants) in the KEYNOTE-158 study. Among the 22 participants with PDA, there were four objective responses (18%), and one complete response, with median duration of response being 13.4 months.59 The study thus supported the use of pembrolizumab for treatment of “previously treated MSI-H/dMMR (DNA mismatch repair) advanced non-colorectal cancer, regardless of anatomic site or origin or tumor histology”.59
Another immunotherapy target is cabiralizumab (MS-986227, FPA008), which inhibits the colony-stimulating factor 1 receptor (CSF1R) and blocks the activation and survival of macrophages. It was recently studied in a phase I study for patients with locally advanced pancreatic cancer or metastatic PDA, in combination with nivolumab.60 The phase I data were exciting, but the phase II data are negative, and the company is not pursuing further investigation.61 While it is an interesting concept to target CSF1R, and the potential use of immunotherapy in PDA, most studies have been unrevealing. We will need to think about other potential combinations at this time. For example, immune checkpoint inhibitors are being tested in combination with other antibodies, such as with pidilizumab (which targets Notch ligand delta-like 1).62 Data are pending.
The final frontier in targeted therapy
Of the four classic mutations in PDA, KRAS has been the elusive holy grail of targeted therapy. RAS genes (HRAS, KRAS, and NRAS) are the most commonly mutated genes in human cancer.63 Over 90% of PDAs have KRAS mutations, making pancreatic cancer “the most RAS-addicted of all cancers”.63 The KRAS protein of the RAS/MAPK pathway, has been deemed “undruggable” due to its smooth 3D structure which lacks good binding pockets for targeted therapy molecules.64 Downstream targets in the RAS/MAPK pathway, such as PI3K/AKT/mTOR, and RAF/MAPK/ERK have also been attempted as targets, without success.64,65 Amgen’s new KRAS G12C inhibitor, AMG-510, unfortunately does not target the specific KRAS mutation typically seen in PDA, but gives hope to the potential of targeting KRAS across multiple cancer types.66
Selected future efforts
Other notable work in the personalization of PDA treatment includes work on patient-derived organoids, which are samples of patient tumor used as preclinical models to evaluate which chemotherapies and targeted therapies are effective in vitro, thus hopefully predicting in vivo activity as well.67 This modeling of a tumor sample may also benefit targeted treatment by mirroring the development of drug resistance “in tandem” with clinical events.67 Research with patient-derived orthotopic xenografts (PDOX) found that the PDOX models kept the same genetic alterations and histopathological features of their primary tumor, and the PDOX-derived organoids’ response to gemcitabine correlated with patients’ clinical outcomes in vivo.67
An exciting concept is that of a “vaccine” for pancreatic cancer. Peptide-based vaccines such as those for telomerase vaccination,68 personalized cancer antigen tolerance preventing vaccines,69 and vaccines against TGF70 have been tried but have not yet come to clinical fruition. Similarly, the GVAX concept of a vaccine is based on developing genetically engineered tumor cells that will release granulocyte-macrophage colony-stimulating factor, but the most recent phase III trial (IMPRESS) did not meet its primary endpoint and thus further development has been stalled.71
Efforts in targeting drug resistance are also important in that they can help maximize the use of current treatment options. Cytosolic 5′-nucleotidase 1A, is a gemcitabine-inactivating enzyme, which works by dephosphorylating gemcitabine monophosphate. It is expressed in tumor cells from resected samples, indicating a potential target for improving and lengthening chemotherapy response.72
Conclusion
In the near future, the majority of cancer-related deaths will be from pancreatic adenocarcinoma.1 Therefore, innovative drug development in PDA is crucial to extend survival for these patients. As we better understand the molecular characteristics and mutations in PDA oncogenesis, improvements can be made in developing targeted treatments based on individual tumor characteristics. While chemotherapy remains the backbone of treatment for this devastating disease, there is hope that targeted therapies and immunotherapies can synergize with current standards of care to improve outcomes for patients with PDA.