As is the case with most solid tumors, gastrointestinal (GI) tumors are treated in a variety of modalities, which are used singularly or in combination, including surgery, targeted therapies, radiation, and chemotherapy. Survival rates by stage in colon and rectal cancer are given inTable 1.
Cancer stem cells (CSCs) are believed to be malignant cells that have the capacity to initiate and maintain tumor growth and survival.1 They are more resistant to radiation and chemotherapeutic agents than other cancer cells.2 The presence of CSCs following cancer therapy may explain the initiation of metastasis and later recurrence of cancers, which can occur even when there is a good initial response to radiation or chemotherapy.3 The first experiments suggesting the presence of CSCs in GI cancers were conducted in 2007.4,5 The investigators used flow cytometry to isolate CSCs using CD133 as a marker and then demonstrated the ability of these CD133-positive cells to form xenografts in non-obese diabetic/severe combined immunodeficiency mice.
Origin of cancer stem cells of the gastrointestinal system
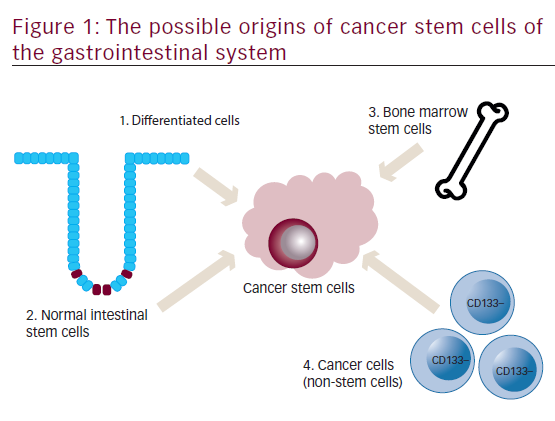
A schematic representation of an individual colon crypt is depicted in Figure 1. Stem cells lie at the bottom of the crypt and through asymmetric division are responsible for generating all epithelial cell types along the crypt–villus axis. GI stem cells may be intrinsically prone to forming CSCs because of their long lifespan combined with rapid turnover and it is widely believed that CSCs are derived from normal stem cells.6,7Other possible sources for GI CSCs include dedifferentiated intestinal cells, possibly via nuclear factor-kappa-B (NF-κB) modulation of Wnt signaling7,8 and bone marrow-derived progenitor cells progressing through metaplasia and dysplasia to cancer.9
Cancer stem cell biomarkers
The CD133 molecule, which is also known as prominin-1, is a pentaspantransmembrane glycoprotein that has been shown to be located mainly in membrane protrusions.10 It was first identified as a surface protein marker of a subset of hematopoietic stem and progenitor cells as early as 1997,11 but its biologic function has yet to be elucidated. In 2007, CD133-positive cells separated from colorectal tumor cells were demonstrated to possess self-renewal properties and high tumorigenic potential.4,5Systematic reviews have indicated that CD133 is a prognostic factor in colorectal cancer (CRC)12 and gastric cancer.13 Carbon nanotube-conjugated CD133-

positive monoclonal antibodies led to photothemolysis of CD133-positive glioblastoma cells in vivo and in mice,14suggesting that CD133 may represent a useful target to selectively inhibit CSCs in CD133-expressing tumor types.
CD44
CD44, a cell surface adhesion molecule, is the principal receptor for hyaluronate, which is the most abundant extracellular matrix component.15,16 CD44 has been suggested to perform functions in CSCs such as mediation of adhesion and homing to the stem cell niche, enhancement of antiapoptotic proteins and surface efflux pump expression, regulation of the cellular redox status and the response to the activation of the canonical Wnt pathway.17–19 Future research is needed to elucidate the suitability of CD44 as a CSC marker in GI cancer and its role in tumorigenesis.
EpCAM
Epithelial cell adhesion molecule (EpCAM), initially described in human CRC as a tumor-associated antigen,20 is expressed highly in a range of human epithelial normal and cancer tissues, including the colon.21 Several lines of evidence indicate that EpCAM is involved in cell adhesion, proliferation, migration, and cancer and stem cell signaling.22,23
Other potential markers
More recently, identified possible markers of CSCs include: CD29/integrin β1, a mucin-like cell adhesion molecule;24 CD24/HSA, a extracellular matrix protein receptor that is involved in regulation of cell migration, proliferation, survival, differentiation, and death;25 Lgr5/Gpr49, a receptor for R-spondin proteins;26and CD166/ALCAM, a cell adhesion molecule.27
Limitations of cancer stem cell markers
Identification and isolation of CSCs using putative surface markers has received much attention in cancer research. However, heterogeneity among GI tumors and GI tumor subtypes has led to difficulty in pinpointing unique markers. Expression of surface markers varies at different tumor stages and their main regulatory functions are not understood fully.28 Lack of universal expression of surface markers has obfuscated their use and no optimal combination of markers has been confirmed for the identification of CSCs. Further, non-CSCs have been shown to also express some of these markers.29 CSC markers are an area of continuous development as more studies identify molecules that may serve as new CSC markers and help to identify CSCs in GI cancers in a tissue-specific manner.
Cancer stemness and epithelial-to-mesenchymal transition
Stemness, initially taken to mean expression of stem cell genes, such as Nanog, Oct4, and Sox2, is a defining property of embryonic and adult stem cells.30Stemness can be measured by a cell’s ability to form spheres when cultured in stem cell media.31 Chemotherapy32 and radiation33 have been found to induce the expression of stemness genes in cancer cells in vitro, thereby enriching the CSC population in the residual tumor.
Epithelial-to-mesenchymal transition (EMT) is the capacity of epithelial cells to acquire mesenchymal traits to allow local invasion into surrounding tissues and systemic dissemination to distant organ sites.3 Recent evidence indicates that EMT can induce differentiated cancer cells into a CSC-like state, suggesting a functional link between stemness and EMT.34
Stemness pathways associated with cancer stem cells in gastrointestinal malignancies— therapeutic targets
The desired aim is to overcome resistance to chemotherapeutics and reduce therapy-related toxicity by developing treatments that are specific for CSCs and that are not toxic to healthy tissues. Examples of stemness pathways as therapeutic targets are discussed below.
Nanog
Nanog, an essential regulator of embryonic stem cell self-renewal that inhibits differentiation, is overexpressed in a variety of cancers including
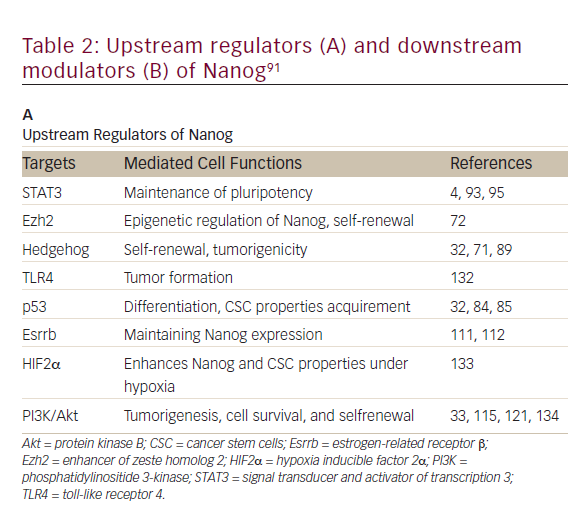
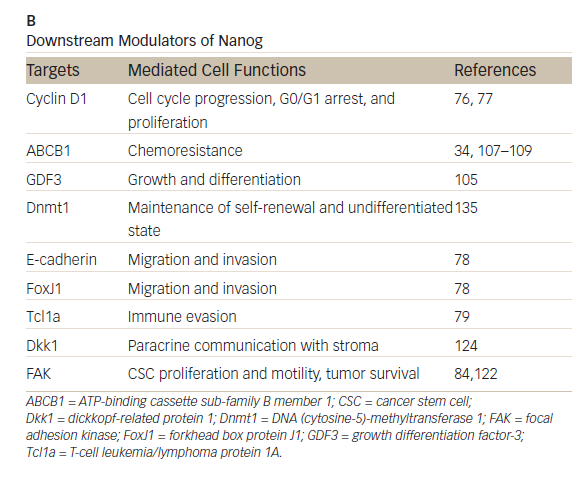
those of the GI system35,36 High levels of Nanog expression are associated with advanced stages of cancer and a poor prognosis, suggesting that it may play a key role in tumor transformation, tumorigenesis, and tumor metastasis.35 Nanog is involved in a complex regulatory network that determines cell fate, proliferation, and apoptosis (Table 2). It is therefore a promising therapeutic target. Genetic ablation of Nanog in SW620 colorectal carcinoma cells suppressed both tumor growth in athymic nude mice and cell proliferation in vitro.37Further, ablation mediated by short hairpin RNA decreased the expression of core CSC transcription factors, supporting a role for Nanog as a signali ng hub in CSCs.38
BBI503 is an orally administered investigational agent designed to inhibit Nanog and other CSC pathways by targeting kinases. Early signs of antitumor activity have been observed in pre-treated patients with advanced cancer (BBI503-101, NCT01781455).39In this ongoing, first-in-human, open-label phase I dose-escalation study, 11 of 20 evaluable patients (55%) had stable disease with a median time to progression of 16 weeks. Of these 11 patients, tumor regression and/or prolonged stable disease
(≥16 weeks) were observed in 10 patients (50% all enrolled patients). Further, in biopsied tumor tissues, dose-dependent pharmacodynamics effects of decreased expression level of Nanog were observed. The recommended phase II dose of continuous once daily BBI503 was 300 mg/day. At this dose, BBI503 was shown to be well tolerated and the pharmacokinetic profile supports once-daily dosing as an acceptable administration schedule. Mild GI adverse events were observed, including grade 1 and 2 diarrhea, adnominal cramps, nausea, and anorexia. Grade 3 diarrhea was noted in two patients at 450 mg once daily.
In an extension study of BBI503-101, BBI503 was administered to patients with advanced CRC.40 Patients (n=47) with heavily pre-treated CRC were enrolled. The disease control rate in evaluable patients (n=39) with high Nanog expression (biomarker positive) was 56%, whereas in biomarkernegative patients, the disease control rate was 13% (p=0.04). Median overall survival in biomarker-positive patients (intent-to-treat) was 38 weeks compared with 16 weeks in biomarker-negative patients (p=0.089 Log-Rank). BBI503 was well tolerated at the recommended phase II dose of 300 mg once daily; grade 3 adverse events were diarrhea (n=5), fatigue (n=4), nausea (n=1), and weight loss (n=1).
STAT3
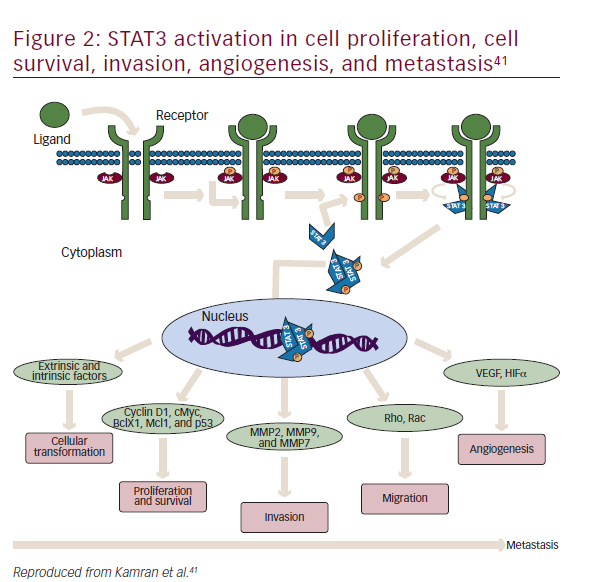
STAT3 is a latent cytoplasmic transcription factor that appears to be activated constitutively in many cancers, to play a pivotal role in metastasis and tumor growth, and is associated with decreased survival.41–43 It was first discovered in 1994 as a signal transducer from cell surface receptors to the nucleus.44 STAT activation has been suggested to be important at every stage of metastasis (Figure 2).
Napabucasin (BBI608) is an orally administered investigational agent that has been designed to inhibit CSC pathways including STAT3, Nanog, and β-catenin pathways by targeting STAT3.41,45 Pre-clinical data indicate that BBI608 inhibits STAT3-driven transcription and suppresses metastasis
and cancer relapse.45,46 An ongoing phase Ib study has demonstrated that BBI608 and weekly paclitaxel can be combined at full dose in patients with advanced malignancies (NCT01325441).47Early anti-tumor activity was shown across several tumor types, especially in those patients with gastric and gastroesophageal junction adenocarcinoma. BBI608-201 (NCT 01325441) is a phase Ib/II study of BBI608 combined with paclitaxel in advanced gastric and gastroesophageal junction adenocarcinoma.48 BBI608 at doses of 480 mg twice daily (BID) and 500 mg BID plus weekly paclitaxel (80 mg/m2) was well tolerated. Common adverse events included grade 1 to 2 diarrhea, abdominal cramping, nausea, and vomiting. Grade 3 adverse events including vomiting, diarrhea of five days’ duration or longer, fatigue, abdominal cramps, nausea, and dehydration. Early signs of anti-cancer activity were observed in a cohort of heavily pretreated patients. The BRIGHTER trial is a phase III randomized, doubleblind, placebo-controlled clinical trial of BBI608 plus weekly paclitaxel versus placebo plus weekly paclitaxel in adult patients with advanced, previously treated gastric and gastroesophageal junction adenocarcinoma (NCT02178956).49 There is also early indication in BBI608-246, that BBI608 can be administered with folinic acid, 5-flurouracil (5-FU) and irinotecan (FOLFIRI) with and without bevacizumab in patients with advanced CRC (NCT02024607),50and in BBI608-224 with panitumumab in KRAS wild-type patients with metastatic CRC (NCT01776307).51 Early signs of anti-cancer activity in these studies warrant further investigation of BBI608. A phase III randomized study of BBI608 plus best supportive care versus placebo plus best supportive care is being assessed in patients with pre-treated advanced colorectal carcinoma (NCT01830621).52
Notch
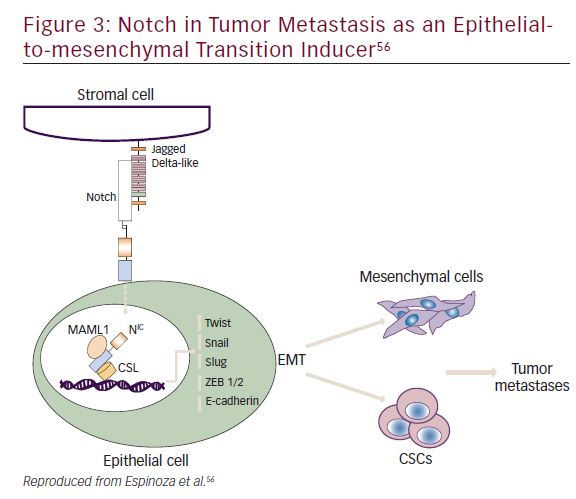
Notch plays a role in embryogenesis, cellular homeostasis, differentiation, EMT, and apoptosis (Figure 3).53–56 Notch signaling is initiated by ligand binding to the Notch receptor, which then undergoes a two-step proteolytic cleavage by a disintegrin and metalloproteases family proteases and γ-secretase.57 Notch signaling can be inhibited by two major classes of Notch inhibitors: γ-secretase inhibitors (GSIs), including RO4929097, MRK-003, MK-0752, and PF03084014, and monoclonal antibodies directing against Notch receptors or ligands. GSIs have been shown to provide clinical benefit; for example, PF0308414 demonstrated early anti-tumor activity in a phase I dose-finding study in patients with advanced-stage solid tumors.58A phase II study of 37 patients with metastatic CRC who had received at least two prior lines of therapy in the metastatic setting found that RO4929097 monotherapy did not demonstrate significant drug activity.59
Notch mediates biologic process through: a canonical pathway (involving ligand-induced cleavage of Notch for transcriptional regulation that includes Notch 1–4 and five Notch ligands Delta like, 1, 3 and 4 and Jagged 1 and 2;60 and non-canonical pathways.55,61 Notch has also been implicated in crosstalk with other oncogenic pathways such as Hedgehog and Wnt signaling.62 Phase I/II trials of Notch inhibitors in solid tumors are in progress (NCT01647828, NCT01859741, NCT01277146, NCT01957007, NCT02259582, NCT01952249, NCT01189929, NCT01952249, NCT01189929). Examples of Notch inhibitors include the compounds tarextumab (OMP-59R5) and demcizumab (OMP- 21M18). OMP-59R5, which blocks both Notch2 and Notch3 signaling, has demonstrated any anti-tumor effect in patient-derived xenograft tumors.63 A phase I trial (n=55) of OMP-21M18, which targets Notch ligand Delta like 4, revealed anti-tumor activity and good tolerability at doses of ≤5 mg weekly.64
Wnt/β-catenin
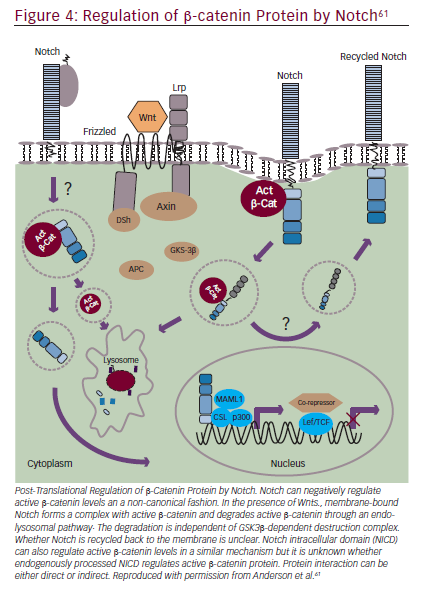
Notch signaling can also interact with Wnt/β-catenin signaling (Figure 4).55,61 The Wnt pathway has a key role in embryogenesis, with effects that regulate proliferation and apoptosis in developing cells.65 Wnt signaling disruptions are observed in a variety of GI cancers66and may have a role in inducing EMT.67 Trials involving Wnt/β-catenin inhibitors are underway, including BBI608 in gastric and gastroesophageal junction cancer (NCT02178956) and in advanced CRC (NCT01776307) as discussed in the STAT3 section. BBI608 has demonstrated early anticancer activity in a phase I trial in patients with CRC and anal squamous carcinoma.68
Monoclonal antibodies, which inhibit Wnt signaling either by neutralizing Wnt ligands or by inhibiting Wnt receptors Frizzled (Fz) and LRP, are also under clinical investigation. Vantictumab (OMP-18R5), a monoclonal antibody that binds five Fz receptors and inhibits Wnt signaling, and a fusion protein decoy receptor, ipafricept (OMP-54F28), are under study in phase I studies in advanced stage solid tumors (ClinicalTrials. gov NCT01345201, NCT02005315, NCT01957007, NCT01973309, NCT01608867, NCT02069145, NCT02092363). OMP-54F28 has been demonstrated to inhibit patient-derived xenograft tumor growth and to decrease CSC numbers.69
Additional signaling pathways
There are many other signaling pathways currently under investigation. Focal adhesion kinase (FAK) is a non-receptor tyrosine kinase70 with important roles in adhesion, survival, motility, metastasis, angiogenesis, lymphangiogenesis, cancer stem functions, tumor microenvironment, and EMT.71–76 Current FAK inhibitors mostly target the FAK kinase domain with the ATP-binding site to inhibit FAK kinase activity.77 Transforming growth factor-β signaling is important for self-renewal and maintenance in the formation of GI cancers.78,79 Hedgehog describes a complex of molecules that regulate cell differentiation, regeneration, and stem cell properties.80 It is central to the development and homeostasis of gut tissue and is deregulated in GI cancers.81The stemness factor Nanog is one of the major targets for Hedgehog signaling.82 Specific inhibitors of Hedgehog signaling, such as vismodegib (GDC-0449), and sonidegib (LDE225) are being examined in clinical trials in addition to the approved indication for basal cell carcinoma.83 Addition of vismodegib to oxaliplatin with 5-FU and folinic acid (FOLFOX) chemotherapy did not improve progression-free survival in patients with gastric and gastroesophageal junction cancer.84
Phosphatase and tensin homolog (PTEN) is a phosphatase that antagonizes the activity of PI3 kinase; PTEN-deficient mice demonstrate an increase in intestinal stem cells, which results in excess crypt formation.85 The PTEN pathway helps regulate the proliferative rate and number of intestinal stem cells.
Induction of cellular quiescence
An alternative to the CSC inhibition discussed involves the chemical induction of cellular quiescence. This is a state of reversible cell cycle arrest, associated with reduced translation rate, activation of autophagy, and a low metabolic rate as characterized by decreased glycolysis.86 This is a burgeoning area of research and it has been suggested that the efficacy of CSC inhibitors and conventional therapies may be enhanced if used in combination with chemoquiescence-inducing agents such as chloroquine and its analogues.87–89
Cancer stem cell inhibitor trial design
Given that the CSCs constitute only a small proportion of an established tumor, a response rate read-out might not be the most suitable endpoint for a CSC inhibitor trial. The effect could be delayed and may better be captured with time-related endpoints using response criteria developed for immunotherapies.90
Conclusion
The discovery of CSCs—which have been described in GI neoplasms such as colon, pancreas, liver, and gastroesophageal tumors—provides promise of a suitable target for improving future oncological treatment. CSCs of the GI system are well suited to research as they are abundant, and have proliferative potential, as well as a uniform structural arrangement which is maintained under tightly controlled signaling pathways. The CSC model has been criticized for failing to take into account the heterogeneous nature of GI cancers. However, CSCs themselves may evolve over time, giving rise to cells that are both genetically and functionally heterogeneous.29Accurate targeting of CSCs must be preceded by precise identification and characterization of those cells. More research is therefore needed to determine which and how many markers need to be considered in the identification of GI CSCs.
CSC-targeted therapies may represent a new treatment modality in patients with cancer. Research continues to target CSCs to render them more chemo- and radio-sensitive, inhibit their potential to proliferate and undergo EMT, thus decreasing the incidence of metastases. Future challenges include optimizing the study design for determining investigational agents’ efficacy in cancer.