Cervical and vaginal cancers remain grave health problems worldwide. While structured screening programs based on the Papinicolaou exfoliative cytology smear have lowered the incidence for invasive disease and thus mortality attributed to cervical and vaginal cancer,1,2 these diseases are still the leading causes of life years lost to cancer in nations poor in health resources.3 The international stage classifications for cancers of the uterine cervix and vagina were revised recently by the International Federation of Gynecology and Obstetrics (FIGO).4,5 Often, cervical and vaginal cancers are lumped together for the purposes of testing therapeutic hypotheses in clinical trials.6 The treatment strategies described in this article are germane to both diseases, with subtleties in radiation therapy and brachytherapy acknowledged in other work.6
Cervical or vaginal cancer disease of bulk greater than 4 cm raises the hazard for disease spread to other pelvic tissues and organs, rendering surgery with the intent to cure challenging. Radiochemotherapy in these scenarios has been met with considerably more success.7–17 Clinical trials have publicized therapeutic gains that occur when radiation sterilizes pelvic disease and is aided in this effort by co-administered chemotherapies with both local and systemic cytotoxic and anticancer biologic effects. Contemporary regimens of radiochemotherapy are discussed for the treatment of women with cervical and vaginal cancers, especially as radiochemotherapy aims to manipulate the ribonucleotide reductase (RNR)-mediated deoxynucleotide triphosphate (dNTP) supply needed for DNA damage repair. Reviewed human translational clinical trials support this notion.
Disrupted Deoxynucleotide Production and Salvage in Cervical and Vaginal Cancers
A primary cause of cervical and vaginal cancers is human papillomavirus (HPV), a virus often acquired by sexual activity.18–21 Many HPVs are associated with anogenital neoplasia, with types 16, 18, 31, 35, 39, 45, 51, 52, 56, and 58 leading to most invasive cancers.18 To hone one’s thinking about radiochemotherapeutic strategies for cervical and vaginal cancers, it is practical to discuss HPV pathophysiology and the means by which the virus initially hijacks cervical and vaginal cell machinery for dNTP production (see Figure 1). HPV contains closed-circular, double-stranded DNA.22 Eight open genome reading frames program early translation of six proteins (E1, E2, and E4–E7) and late translation of two proteins (L1 and L2) for multiplication of viral copy number in cells.22 For viral genome replication, HPV interrupts host-cell mechanisms of cell-cycle termination. In doing so, HPV-E6 binds to the ‘superintendent’ p53 protein, prompts its elimination, and thus lifts molecular barriers halting cell-cycle transit through a G1/S cell-cycle restriction checkpoint.23–25 At the same time, HPV-E7 removes restrictions linked to the retinoblastoma protein through a proteosome-dependent degradation pathway.26 This HPV-E7-effect promotes E2F activation,27 which turns on S-phase replication proteins responsible for normal host-cell gene duplication including subunits of RNR. The far-reaching effects of HPV proteins that supersede cell-cycle checkpoints increase the tendency to oncogenic phenotypes.22 Current HPV biology, therefore, entices investigators to exploit pharmacologic means of limiting 2’-deoxyribonucleoside diphosphate (dNDP) production by RNR (maybe its M1-M2 form specifically) through either transcriptional or protein–protein regulation.
Staying with this motif, the HPV-E6 effect of lowering p53 provides collateral manipulation of RNR. The RNR α2β2 heterotetramer (or its higher order α6β6 or α6β2 forms) rate limits the extraction of a 2’-hydroxyl moiety for hydrogen in a ribonucleoside diphosphate, bringing about a dNDP.28,29 Once phosphorylated, the dNDP becomes a building block for DNA as a 2’-deoxyribonucleoside triphosphate (dNTP). The large subunit α (M1) contains three functional sites: a catalytic site for ribonucleoside substrates, a specificity site, and an activity site that by allosteric regulation,controls specific dNTP output by RNR. The small subunit β (M2 or p53R2 [M2b]) quarters a free radical that shuttles to and from the M1 catalytic site with each enzymatic cycle. The M2 and M2b subunits have been shown to be overexpressed in HPV-related cervical cancers.17
Learning about the manners by which RNR and its enzymatic activity can be manipulated has taught the oncologist why anticancer strategies may be or may not be successful in cervical and vaginal cancer disease management (see Figure 1). Proton-coupled electron transfer from the M2 or M2b diferric tyrosyl radical (•Y122) through a 35 Å amino acid electron and proton tunnel to the catalytic site on M1 (cysteine C439) forms the root mechanism by which RNR accomplishes its task of catalyzed ribonucleotide reduction. As worked out in Escherichia coli, an amino acid chain of •Y122/W48/Y356 in R2 and then Y731/Y730/C439 in R1 facilitates radical transport.30 It is substrate turnover in the catalytic site of M1 initiated by a thiyl radical (C439) that abstracts a hydrogen from the ribose ring of ribonucleotide diphosphates to generate dNDPs.30 A complementary salvage of deoxynucleosides (dNs) to dNMPs and ultimately dNDPs has also been shown (see Figure 131,32). Both the creation of and the recapture of deoxynucleosides are very narrowly regulated because unbalanced dNTP reserves result in deleterious genotoxic stress.33 As such, equal manufacturing of dNDPs for DNA replication or DNA repair arises by way of a feedback ladder of allosteric effectors at an M1 specificity site.34 In the feedback ladder, thymidylate synthase provides the sole intracellular source of de novo deoxythymidine monophosphates, and thus the 5-fluorouracil (5FU) blockade of thymidylate synthase modulates allosteric regulation of RNR. Also, cells may regulate RNR M1, M2, and M2b protein levels – by very slow degradation for M1,28 by quick degradation in late mitosis through a KEN-box for M2,35,36 or by p53-dependent transcription or p53 protein–protein binding.37 It is speculated that sources of dNDPs after DNA-damaging insults include ribonucleotide reduction first by a M1-M2b mediated process and subsequently by a M1-M2 mechanism.38–40
Large amounts of dNDPs are needed in cells duplicating their genome, and regulated means of turning on RNR for this task are known. Less is known about how cells meet urgent demands for the repair of damaged DNA after DNA-damaging therapies. Indeed, cells may necessitate fewer dNTPs to fix short stretches of DNA damaged by a cisplatin adduct. However, after ionizing radiation, thousands of altered bases (e.g., 8-oxoguanine) in intracellular RNA, genomic DNA, and mitochondrial DNA, plus about 1,000 single-strand breaks, and plus nearly 40 double-strand DNA breaks are inflicted upon cells. Demand for dNTPs (and thus dNDPs) to mend this damage puts cells in a vital, high-stakes gamble using quick and short-run scalable pathways of dNTP supply. Nature balances such demand–supply economics by utilizing not only a de novo RNR enzyme reduction of ribonucleotides, but also a complementary deoxynucleoside salvage system. For the oncologist, the ability to disrupt the natural demand–supply balance sheet through an increase in dNTP demand (i.e., radiation, cisplatin) while also restricting supply through pharmacologic inhibitors of de novo RNR activity (i.e., gemcitabine, hydroxyurea [HU], and triapine) has been tested in clinical trials. Means of halting deoxynucleoside salvage, and thus ultimately dNTP recovery, remain largely undiscovered and untested.
Radiation Therapy for Cervical and Vaginal Cancer
For cervical and vaginal cancers, radiation remains a cornerstone of therapy. Careful design of a four-field arrangement of anterior, posterior and opposed lateral ports delivers 4,500 cGy in 25 divided daily high-energy (6–18 megavolt) fractions of 180 cGy. Such a regimen is commonly administered for five weeks, typically on a Monday through Friday daily therapy schedule. When done in this way, shielding spares the bowel anteriorly, but comes at a cost of more radiation dose delivered to the iliac wings (and bone marrow), skin, and subcutaneous tissue than would be accomplished with anteroposterior and posteroanterior (AP/PA) opposed high-energy radiation fields. If cancer is felt to involve the lower one-third of the vagina, ports may be opened inferiorly to allow groin-node irradiation. Commonly, a two-field AP/PA parametrial boost adds dose (540–900 cGy) to the lateral pelvis while limiting midline dose to the rectum and bladder to supplement the dose given by brachytherapy. Snapshots of typical radiation fields are given in Figure 2A and B. As an alternative, radiation oncologists have begun to explore the benefits attributed to intensity-modulated radiation therapy (IMRT). Clinical trials are integrating IMRT as a way of radiation delivery because of its favorable gastrointestinal41 and hematologic42 toxicity characteristics. If the efficacy of IMRT is judged ultimately not to be inferior to conventional four-field therapy,43 IMRT perhaps will become more useful. As of yet, IMRT remains an active research area; without further study, IMRT should not be routinely used for women with intact cervical and vaginal cancers.
When there is a desire to treat retroperitoneal para-aortic lymph nodes, an extended-field modification of the four-field arrangement can be done (Figure 2D–2F). One technique uses a lower half-beam four-field arrangement and an upper half-beam AP/PA arrangement. A more modern technique matches the half-beam four-field to an IMRT para-aortic field. This is done in an effort to reduce late consequential gastrointestinal sequelae.44
Brachytherapy implants, where radiation sources are close to the cervical or vaginal cancer targets, can be accomplished by many techniques to add additional dose to the primary cancer (3,500–4,000 cGy). Radiation source selection, often cesium 137 or iridium 192, determines the radiation dose rate, often coined low-dose-rate (cGy per hour) or high-dose-rate (cGy per minute). Intracavitary and interstitial applicators and implantation techniques are quite similar and beyond the scope of this article. The reader is referred to other sources for more detailed discussion.6 The prescription for brachytherapy is Point A, an anatomical spot 2 cm superior and 2 cm lateral to the cervical os in the plane of a uterine tandem. A low-dose-rate brachytherapy example is shown in Figure 2C.
Cervical and vaginal cancers are typically treated with a radiation dose of 8,000 cGy or more. Nowadays, this is accomplished between external beam radiation and brachytherapy, usually happening within an eight-week span, because up to a -1 %/day hazard for lowered disease control has been observed.45
Radiochemotherapy for Cervical and Vaginal Cancer
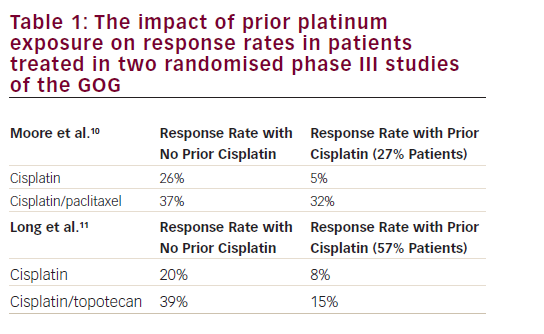
One unifying concept of chemotherapy, as it applies to cervical and vaginal cancer management, is how administered chemotherapy damages DNA or slows its repair. One of the first anticancer strategies for cervical cancer targeted the RNR tyrosyl radical with HU by the Gynecologic Oncology Group (GOG-004).7 In its one-electron reductive state,46 oral HU irretrievably disrupts the M2/M2b diferric tyrosyl radical (•Y122), rendering the electron and proton tunnel to the M1 active site inactive. As it happens, women were assigned by random assortment to either HU (80 mg/kg, oral twice weekly) or placebo during pelvic radiation. Three-year endpoints of pelvic disease control and of progression-free survival (i.e., cancer relapse or death) were better after radiation–HU than radiation–placebo (see Table 1, p<0.05). As one of the first randomized clinical trials of its kind in gynecologic cancer, HU-mediated block of RNR during radiation became the measure of other radiochemotherapeutic agents in cervical and vaginal cancers.
Capitalizing on this experience, GOG investigators tested whether a radiation–cisplatin–5FU combination stood up against a radiation–HU strategy (GOG-085).8 Here, it was thought that 5FU impeded thymidylate synthase, eventually dismantling the dNTP feedback ladder that determined the selectivity site of RNR. Also in this trial, a new chemical agent was introduced for study in cervical and vaginal cancers. In contrast to the almost instaneous damage induced by ionizing radiation, cisplatin renders cytotoxic DNA damage protracted over a 6–24 hour course to keep cells actively engaged in DNA-adduct repair for constant dNTP payouts from RNR.47 Thus, in this randomized clinical trial, women took radiation and HU (80 mg/kg, oral twice weekly) or had radiation, bolus infusion of cisplatin (50 mg/m2 four hours before radiation, days one and 29), and protracted infusion of 5FU (1,000 mg/m2/day, days 2–5 and 30–33). Up to that time, the more common and consequential component of treatment failure was ineffective sterilization by radiation alone. Adding more DNA damage in the form of cisplatin adducts and impairing RNR ability to respond to the demands for dNTPs through 5FU’s skewing of the dNTP feedback ladder, the radiation–cisplatin–5FU treatment resulted in improved disease control (see Table 1) and progression-free survival (p=0.033). In a similar way, investigators from the Radiation Therapy Oncology Group (RTOG-9001) found similar results.9,10 In this trial, conducted contemporaneously with GOG-085, women were allocated randomly either to a treatment arm of extended-field radiation alone or to four-field pelvic radiation, bolus cisplatin (75 mg/m2 within 16 hours of radiation, days one, 22, and 42), and 5FU (1,000 mg/m2/day, days 2–5, 23–26, and 43–46). Compelling data for pelvic disease control, extrapelvic disease control (see Table 1), and three-year progression-free survival (p=0.0003) were documented for the radiation–cisplatin–5FU regimen versus radiation alone regimen.
Soon afterward, investigators were motivated to explore coordinating optimal DNA damage wrought by radiation and by cisplatin with peak pharmacologic RNR blockade. First, the GOG conducted a randomized trial (GOG-0043) of three cisplatin dose schedules in 496 women for persistent pelvic or extrapelvic cervical cancer disease response. The majority (93 %) of women had received pelvic irradiation at a time prior to receiving cisplatin infusions. Regimen 1 was cisplatin 50 mg/m2 every 21 days, regimen 2 was cisplatin 100 mg/m2 every 21 days, and regimen 3 was cisplatin 20 mg/m2 daily for five consecutive days. Regimens were repeated for a total ceiling dose of 400 mg/m2 providing there was no evidence of tumor progression. In the 444 evaluable patients for responses attributable to cisplatin’s DNA-damaging effect, the sums of complete and partial response rates were 21 % for regimen 1, 31 % for regimen 2, and 25 % for regimen 3 (p=0.015). With a greater appreciation that cisplatin was cytotoxic on its own, had an ability to synchronize cells in radiosensitive phases of the cell cycle, and contributed damage that would tax dNTP supply, investigators sought out clinical trials of single-drug and multiple-drug combinations with radiation for the treatment of cervical cancer.
One clinical trial (GOG-120) in women with cervical cancer allotted, by random selection, one of three cisplatin-based radiochemotherapy treatments.11 Regimen 1 was daily pelvic radiation plus weekly cisplatin (40 mg/m2) followed by brachytherapy. Regimen 2 was day one and day 29 bolus cisplatin (50 mg/m2) plus 96 hour infusion of 5FU (4,000 mg maximum) plus oral HU (twice weekly, 2,000 mg/m2) during radiation and followed by brachytherapy. Regimen 3 was twice-weekly oral HU (3,000 mg/m2) during radiation and followed by brachytherapy. Three-year pelvic disease control and extrapelvic disease control was found to be superior with regimens 1 and 2 (see Table 1). Mixing two RNR inhibitors, HU and 5FU, contributed no added clinical benefit over the weekly cisplatin regimen (see Table 1). Long-term follow-up confirmed a progression-free survival advantage for regimen 1 (p<0.01) and regimen 2 (p<0.01), as compared to regimen 3.12 For ease of clinical administration, the weekly cisplatin regimen was adapted quickly to radiochemotherapy practice.48 To confirm the results in an independent trial, investigators then randomized women with cervical cancer to weekly single-agent cisplatin (40 mg/m2) versus single-agent 5FU (protracted venous infusion 225 mg/m2/day, days 1–5 of a five weekly cycles).15 The superiority of cisplatin was verified (see Table 1).
The ability of radiochemotherapy to effect sterilization of primary cervical cancer has been interrogated in actual surgical specimens.14 It was hypothesized that through an analysis of extrafascial hysterectomy histopathologic samples from two randomized clinical trials (GOG-071, GOG-123) would provide in vivo proof-of-principle evidence of a radiation and a radiochemotherapy effect.13,49 Among 464 hysterectomy samples, a significantly higher proportion of a good response (80 %, greater than 90 % sterilization) was observed after radiochemotherapy as compared to radiation alone (71 %, p<0.037). Efforts to improve primary disease have re-investigated the role of pharmacologic inhibitors of RNR.
To be pharmacologically useful, nucleoside analogs, such as cytarabine, fludarabine, and gemcitabine, require activation by deoxycytidine kinase in the salvage pathway of deoxynucleosides.50 Of these, gemcitabine annihilates the M1 catalytic site of RNR by a covalent bond when it appears as a cytidine diphosphate analog, thereby destroying both M1 subunits of a RNR α2 dimer.51 Phase 2 data of radiation, cisplatin (40 mg/m2), and gemcitabine (125 mg/m2) provided a 78 % complete response rate.52 In a phase 3 cervical cancer patient trial, regimen 1 consisted of once weekly cisplatin (40 mg/m2) plus gemcitabine (125 mg/m2) during pelvic radiation followed by two adjuvant 21-day cycles of cisplatin (50 mg/m2, day one) plus gemcitabine (1,000 mg/m2 on days one and eight). Regimen 2 involved once weekly cisplatin (40 mg/m2) and the same radiation.16 The hazard for relapse or death was reduced by 32 % when gemcitabine was included in the treatment (see Table 1, p=0.023). However, conflicting interpretations of this data emerged53 Gemcitabine radiochemotherapy may have been successful because it cured more women upfront from its radiosensitizing effect. Or else, gemcitabine radiochemotherapy may have been successful because it delayed scoring of disease relapses from an adjuvant lead-time bias effect. Either interpretation stands from the data reported. Also, in a GOG effort (GOG-9912), this regimen was deemed too toxic.54 Further study is warranted.
Attacking RNR at its M2/M2b diferric tyrosyl radical (•Y122) with the more potent RNR inhibitor 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) has also been investigated. The putative mechanism here is a molecular interaction of an Fe2+–3-AP chelate and of oxygen generating local reactive oxygen species capable of annihilating the nearby tyrosyl free radical.55 As such, timed after DNA damage (e.g., damage promulgated by ionizing radiation), 3-AP’s cell death-provoking effect may be due to its inability to supply on-the-spot dNTPs needed for DNA damage repair.39,40 A phase 1 clinical trial has been conducted in 10 women with cervical cancer involved intravenous 3-AP (25 mg/m2, three times weekly) plus once-weekly cisplatin (40 mg/m2) during daily radiation.17 Median survivor follow-up is now 38 months and progression-free survival is 88 % at three years (see Table 1). A phase 2 study of 3-AP-cisplatin radiochemotherapy in 25 additional women with cervical and vaginal cancers has completed enrollment, with premature response data indicating comparable clinical advantage.
Conclusion
In an age of biologic anticancer therapies, women with cervical and vaginal cancers have profited from improved appreciation of cellular DNA-damage responses to radiochemotherapy. Alternative means of RNR inactivation have provided gains in radiochemotherapeutic response as well. Clinical research stakeholders, both patient and physician, need to advocate for oral or transdermal radiosensitizing therapies that can be combined safely with radiation so that women may receive effective treatment equally in resource-rich and impoverished nations. Novel agent scheduling, patient pre-existent morbidity, and treatment-related toxicity are recognized mitigating factors in the clinical development of helpful anticancer treatment regimens.56 Leaps forward in the radiochemotherapeutic management of cervical and vaginal cancers will likely include cytotoxic agents that induce DNA damage paired with biologic agents that slow or halt its repair. ■