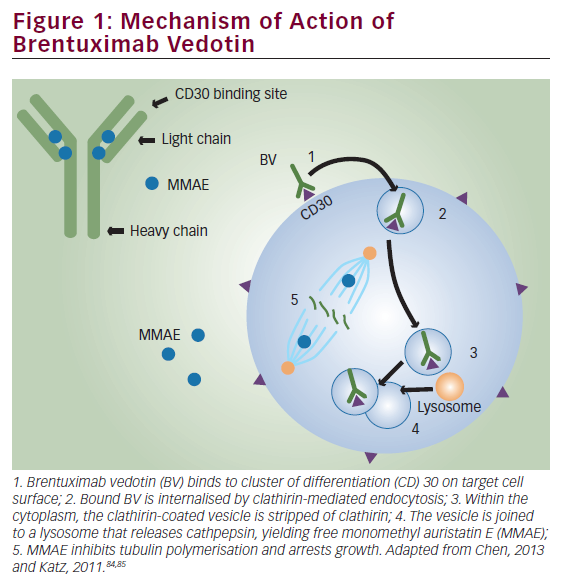
The addition of rituximab to chemotherapy provides an overall survival benefit in aggressive lymphomas,1 whereas for indolent lymphoma subtypes this addition has resulted in improvements in progression-free survival.2 For more than two decades, immunotherapy with monoclonal ‘naked’ antibodies targeting CD20 remained the primary representative of this therapeutic modality,3 with a small number of patients receiving anti-CD20 radioimmunotherapy.4
A number of immunotherapeutic agents have been developed in the past 5 years, most targeting new cell surface antigens. Targets include CD79a and CD19. CD79a is a central component of the B-cell receptor, which is targeted by the currently approved antibody–drug immunoconjugate, polatuzumab vedotin.5 CD19 is a ‘pan B’ surface marker present throughout B-cell maturation stages and has long been studied as a potential therapeutic target.6–8 Multiple approved agents targeting CD19 are now available, including chimeric antigen receptor (CAR) T cells, monoclonal antibodies (mAbs) and antibody–drug conjugates (ADCs). While all three therapeutic modalities are aimed at the same target, they have unique mechanisms of action, adverse safety profiles and indications.
CD19 as target in lymphoma
Biology of CD19
CD19 is a 95 kD glycoprotein member of the immunoglobulin superfamily. It is composed of a highly conserved cytoplasmic domain, a single transmembrane domain and two extracellular C2 immunoglobulin-like domains separated by a small non-Ig-like helical region.9 The expression of CD19 throughout B-cell maturation stages, from late pro-B cells or early pre-B cells until loss of expression during terminal plasma cell differentiation, make it a reliable marker for normal and neoplastic B cells.9,10 Initial CD19 expression occurs at the time of immunoglobulin heavy chain gene rearrangement10,11 and is tightly regulated throughout B-cell maturation, with mature B cells having 3-fold higher expression,9 reported to range between 10,000 and 100,000 sites per cell.12,13 CD19 forms part of a multimolecular signalling complex in the surface of the B cell along with CD21, the tetraspanin membrane protein CD81, and CD225.9,10,14 Additionally, CD19 modulates/amplifies B-cell receptor-dependent and -independent signals through activation of Src family kinases as well as PI3K and AKT.
Once mAbs (or antibody conjugates) bind to B-cell surface antigens, they undergo internalization and trafficking to lysosomes, where the antibody is degraded and a conjugated payload (toxin, cytotoxic agent or radioisotope) is released into the cytoplasm.12,15 Press et al. described several characteristics of mAbs relevant for the choice of ‘antibody-directed immunotherapy’, including target antigen density and tissue specificity, antibody immunoreactivity, avidity, isotype, rate of degradation and intracellular routing.12 Perhaps persistence of surface antigen should be included as a relevant characteristic. Anti-CD19 antibodies were noted at the time to have high antigen density (although lower than anti-CD20), rapid internalization (slower than anti-CD22 and faster than anti-CD20) and adequate internal routing and antigen persistence. These characteristics suggested CD19 as a valuable target for antibody-based immunotherapy.
Immunotherapy targeting CD19
Initial studies
Early studies with mAbs targeting CD19 for treatment of B-cell malignancies showed limited clinical activity as single agents16 or in combination with interleukin-2.17 Subsequently, an antibody–toxin conjugate of anti-B4-blocked ricin (anti-B4-BR) as post autologous stem cell transplant maintenance for patients in complete remission showed that 23/31 infused patients developed human anti-mouse or anti-ricin antibodies and a continued relapse pattern was observed.18
Optimized monoclonal antibodies
Subsequent developments of immunotherapy included the development of Fc-engineered mAbs, CAR T cells and ADCs. Among Fc-engineered mAbs, inebilizumab (MEDI-551) is a humanized, affinity and afucosylated IgG1 kappa mAb targeting CD19, with enhanced antibody-dependent cellular cytotoxicity (ADCC).19 Phase I results suggested responses in aggressive and indolent lymphomas,20 but combination studies and randomized phase II trials showed limited activity.6 Tafasitamab (MOR208, XmAb5574) is an Fc-engineered domain with improved binding to Fcγ receptors21 and increased ADCC.21,22 Single-agent activity of tafasitamab against aggressive and indolent lymphomas was limited (overall response rate [ORR] for diffuse large B-cell lymphoma [DLBCL] 26%, follicular lymphoma [FL] 30%, chronic lymphocytic leukaemia 22%) and consisted mostly of partial responses (PR).23 L-MIND (A Study to Evaluate the Safety and Efficacy of Lenalidomide With MOR00208 in Patients With R-R DLBCL; ClinicalTrials.gov identifier: NCT02399085) was a phase II trial evaluating the combination of tafasitamab and lenalidomide in relapsed or refractory (r/r) DLBCL in transplant-ineligible patients. The combination resulted in an ORR of 60%, with a complete response (CR) in 43% and a median duration of response of 43.9 months.24,25 These results led to regulatory approval of tafasitamab in combination with lenalidomide for treatment of patients with r/r DLBCL after ≥1 line of therapy who are ineligible for autologous stem cell transplant.
Bispecific antibody constructs
Bispecific antibody constructs correspond to a large group of molecules that includes immune molecules capable of recognizing two distinct epitopes.26 The formats can be variable in size and structure, from small proteins with linked antigen-binding fragments, to large multidomain proteins. While antigen selection may be varied, the bispecific agents targeting CD19 are mostly T-cell engagers, targeting CD3 as the second epitope.26 Blinatumomab is a bispecific T-cell engager that is approved for the treatment of CD19-positive acute lymphoblastic leukaemia. In the initial phase I trial of blinatumomab, neurological toxicity was common (all grades 71%, grade 3 22%). Among patients treated at the maximum tolerated dose (60 µg/m2/day), responses were observed across several lymphoma subtypes (FL 12/15, DLBCL 6/11, mantle cell lymphoma [MCL] 5/7) although, again, most were PRs.27 In a subsequent phase II trial, dose increase and steroid premedication over the course of 3 weeks permitted escalation of blinatumomab to higher doses (112 µg/m2/day), with lower incidence of neurological toxicity. In addition to neurological toxicity, the biggest challenge of blinatumomab is its pharmacokinetic profile, with a very short half-life requiring continuous infusion for 4–8 weeks. Moreover, dose increases or re-initiation of infusion after an interruption require inpatient monitoring. These logistical challenges have limited further development of blinatumomab in non-Hodgkin lymphomas.
Chimeric antigen receptor T cells
CAR T cells are cells that have been genetically modified to express a chimeric receptor that contains an extracellular B-cell antigen recognition with transmembrane and intracellular domains than mediate T-cell activation. Four anti-CD19 CAR T-cell therapies are currently approved for treatment of r/r B-cell malignancies: axicabtagene ciloleucel for DLBCL and FL,28 tisagenlecleucel for acute lymphoblastic leukaemia and DLBCL,29,30 lisocabtagene maraleucel for DLBCL31 and brexucabtagene autoleucel for MCL. ORRs in DLBCL range between 52% and 83%, with CR rates of 40–54%. Long-term remissions are achieved in approximately 40% of r/r DLBCL cases. The adverse event pattern of these adoptive immune therapies appears to be consistent, with acute immuno-inflammatory toxicities including cytokine release syndrome and immune effector-associated neurotoxicity syndrome being the predominant complications. Prolonged cytopenia, hypogammaglobulinaemia and opportunistic infections have also been reported.28–33 The incidence of adverse events appears to be slightly different between CAR T constructs containing CD28 versus 4-1BB co-stimulatory signals.
Antibody–drug conjugates targeting CD19
ADCs are antibodies bound to other molecules through a chemical linker. The ‘payload’ can include small potent cytotoxic molecules, protein toxins, immunomodulatory proteins, radionuclides and others. ADCs can be considered site-specific prodrugs.34 As discussed above, the internalization kinetics of CD19 make it an attractive target for these agents,12 as internalization and lysosomal degradation are the mechanisms through which the payload is released to the target cell population.
Several ADCs targeting CD19 have been evaluated in clinical trials, including coltuximab ravtansine, denintuzumab mafodotin, SGN-CD19B and loncastuximab tesirine. Coltuximab ravtansine (SAR3419) and denintuzumab mafodotin (SGN-CD19A) were ADCs conjugated to antimicrotubule agents, specifically a maytansinoid and monomethyl auristatin F. Both agents were associated with ocular toxicity: 44% for coltuximab and 65% for denintuzumab.35 Blurred vision was the most common manifestation and corneal epitheliopathy was the main ocular finding.35 A phase II trial of coltuximab failed to meet its primary endpoint, achieving responses in only 31% of patients, most of them PRs.36 Further development of both agents has been discontinued.37 Other anti-CD19 activity in preclinical models includes SGN-CD19B (pyrrolobenzodiazepine [PBD] payload), HuB44-DGN452 (alkylating agent payload)38 and CD19-thiomab-Amanitin (RNA polymerase II inhibitor payload).6,39
Loncastuximab tesirine
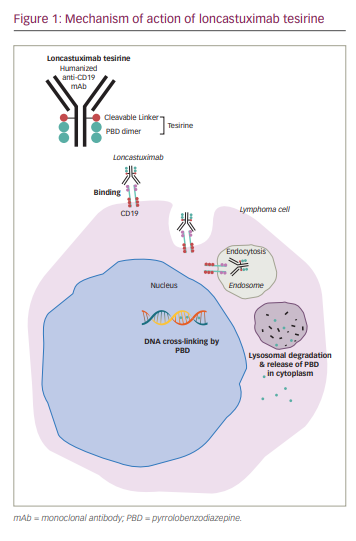
Loncastuximab tesirine is an ADC comprising SG3199, a PBD dimer, conjugated to a humanized IgG1 anti-CD19 mAb through a valine-alanine protease cleavable linker.
PBD as a monomer is derived from Actinomyces and has potent alkylating properties.40 Early ADC development conjugated a synthetic PBD dimer payload, talirine (SGD-1910), with a CD33 mAb for the treatment of acute myeloid leukemia.41 Tesirine (SG3249) was developed through the addition of a PEG8 spacer, incorporation of N10 nitrogen in the cleavable valine-alanine prodrug linker, and substitution of C2 aryls to methyl groups in the talirine structure. These modifications were aimed at increasing solubility without decreasing potency. The substitution of the C2 aryls with methyl groups allowed for more flexible and discrete inter-strand DNA binding that evades the host DNA repair mechanisms.40,42
Preclinical studies show that loncastuximab tesirine is internalized after binding to surface CD19 followed by release of the PBD payload through lysosomal cleavage of the protease linker, resulting in neoplastic cell cytotoxicity (Figure 1).42,43 Animal studies measured the cytotoxic effects on CD19+/CD19- tumours after administering free SG3199 warhead and loncastuximab tesirine. The preclinical studies showed dose-dependent antitumour activity of loncastuximab tesirine. In addition, the investigators observed bystander killing of CD19-negative cells, considered helpful in targeting tumours with decreased expression of CD19.43
Clinical trials
A single-arm, multicentre phase I trial with single-agent loncastuximab tesirine included 183 patients with r/r B-cell lymphoma (Table 1).44,45 The dose-escalation phase included 88 patients and the dose-expansion phase included 95 patients. The diagnoses included DLBCL (n=139), FL (n=14), MCL (n=15) and other histologies (n=15). The median number of prior therapies was 3. Loncastuximab tesirine was given intravenously over 60 minutes on Day 1 of each 21-day cycle at doses of 15–200 µg/kg. No dose-limiting toxicities were observed and none of the dose levels met maximum tolerated dose criteria. However, cumulative toxicity was observed at 200 µg/kg, with higher incidence of treatment-emergent adverse events (TEAEs) compared with the 120 and 150 µg/kg cohorts. These two levels were selected for the dose-expansion cohorts (120 µg/kg n=26; 150 µg/kg n=69). The median number of loncastuximab tesirine cycles was 2 (range 1–24), with a median dose per cycle of 129.9 µg/kg. The median duration of treatment was 64 days (range 22–532 days). A total of 181 patients (98.3%) experienced at least one TEAE. Haematological toxicity was common, including thrombocytopenia, anaemia and neutropenia. Among non-haematological TEAEs, the most common were fatigue (n=78, 42.6%), nausea (n=59, 32.2%), peripheral oedema (n=58, 31.7%) and elevated gamma-glutamyl transferase (GGT; n=57, 31.1%). Cutaneous reactions were also common, including rash in 45 patients (24.6%), most frequently in sun-exposed areas. Oedema and effusions occurred in 86 (47.0%), including both peripheral oedema (n=58, 31.7%) and pleural effusions (n=39, 21.3%). The incidence of oedema and effusions decreased after the introduction of corticosteroid premedication. Grade 3 and higher TEAEs were observed in 77% of patients. The most frequent grade ≥3 adverse events were haematological, followed by elevated GGT and electrolyte abnormalities. Progressive disease was the primary cause of treatment discontinuation (n=83, 45.4%).
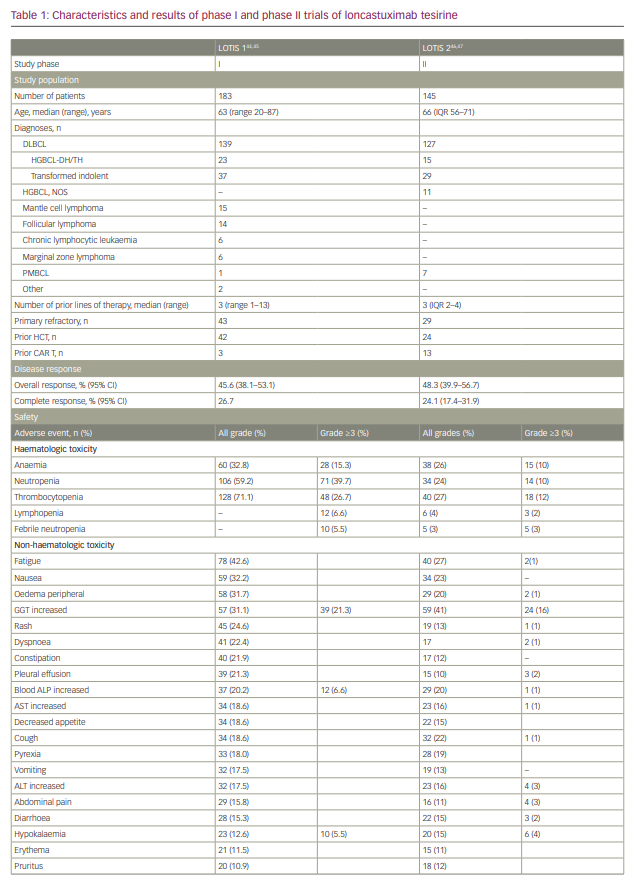
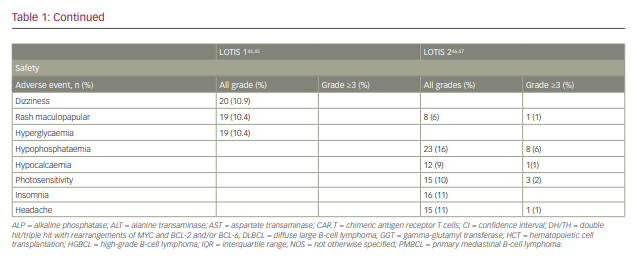
The ORR in the evaluable population (n=180) was 45.6% (95% confidence interval 38.1–53.1%), with 26.7% showing a CR. A dose–response relationship was observed, with ORRs of 29.4% for patients treated with doses of 15–90 µg/kg compared with 47.2% for patients treated at doses of 120–200 µg/kg.
Immunogenicity analysis found anti-drug antibodies were infrequent and with low or very low log2 titres, suggesting loncastuximab tesirine did not induce the formation of anti-drug antibodies.
LOTIS-2 (Study to Evaluate the Efficacy and Safety of Loncastuximab Tesirine in Patients With Relapsed or Refractory Diffuse Large B-Cell Lymphoma; ClinicalTrial.gov identifier NCT03589469) was a multicentre, phase II trial that enrolled patients with r/r DLBCL after two or more prior lines of therapy (Table 1).46 Loncastuximab tesirine was given intravenously every 3 weeks with an initial loading dose of 150 µg/kg for the first two cycles, then 75 µg/kg for subsequent cycles for a maximum of 12 months. A total of 145 patients were enrolled. The median number of cycles was 4. ORR was 48.3%, with 24.1% of patients achieving CR.46 The median duration of response for the entire population was 13.4 months. Subgroup analyses showed comparable response rates and duration of response in specific subgroups, including elderly patients, and those with high-grade lymphoma with MYC and BCL-2 and/or BCL-6 rearrangements, transformed indolent lymphoma, and primary refractory disease.47 The primary causes of treatment discontinuation were progressive disease and treatment intolerance. The most frequent all-grade TEAEs included elevated GGT (40.7%), neutropaenia (39.3%), thrombocytopaenia (33.1%), fatigue (27.6%) and anaemia (26.2%). Among grade ≥3 TEAEs, the most common were neutropaenia (25.5%), thrombocytopaenia (17.9%), elevated GGT (16.6%) and anaemia (10.3%). Tolerability was similar among all age groups.47
Ongoing combination studies
The single-agent activity and specific safety profile of loncastuximab tesirine make it an attractive agent for combination with other drugs. LOTIS-3 is a phase I/II trial (NCT03684694) evaluating the safety and recommended dose and schedule of loncastuximab tesirine in combination with the Bruton’s tyrosine kinase inhibitor, ibrutinib.48 The target population includes patients with r/r DLBCL and MCL. The dose-escalation phase of the study is complete. The dosing on the expansion phase includes loncastuximab tesirine given in cycles 1 and 2 at a dose of 60 µg/kg, followed by ibrutinib alone in cycles 3 and 4. Loncastuximab is then repeated in cycles 5 and 6 in responding patients. Ibrutinib is given at a dose of 560 mg for up to 1 year. In a recent update, 37 patients were enrolled (30 DLBCL, 7 MCL). The median number of prior therapies was 2 (range 1–6) and 18 patients were refractory to the last line of treatment. The median number of cycles of loncastuximab tesirine and ibrutinib were 2 (range 1–4) and 4 (range 1–14), respectively. Median treatment duration was 105 days (range 18–379). All patients experienced TEAEs, with haematological toxicity including thrombocytopaenia (29.7%) and anaemia (24.3%) being most common. Non-haematological toxicity included fatigue, rash and diarrhoea (all 21.6%). Grade ≥3 events occurred in 24 patients (64.9%), most frequently haematological in nature. The ORR and CR rate were 63.9% and 36.1%, respectively. This study is ongoing with enrolment currently in the phase II portion.
Upcoming trials
LOTIS-5 (Study to Evaluate Loncastuximab Tesirine With Rituximab Versus Immunochemotherapy in Participants With Relapsed or Refractory Diffuse Large B-Cell Lymphoma; ClinicalTrials.gov indentifer: NCT04384484) is a phase III, randomized controlled study enrolling patients with r/r DLBCL and comparing loncastuximab tesirine plus rituximab versus rituximab, gemcitabine and oxaliplatin. Estimated enrolment is 350 patients.49 An investigator-initiated phase II trial is evaluating the combination of loncastuximab tesirine and rituximab for treatment of r/r FL (NCT04998669),50 whereas LOTIS-6 (Study to Evaluate the Efficacy and Safety of Loncastuximab Tesirine Versus Idelalisib in Participants With Relapsed or Refractory Follicular Lymphoma; ClinicalTrials.gov identifier: NCT04699461) will be a randomized phase II trial comparing loncastuximab tesirine and idelalisib for treatment of patients with r/r FL.51 LOTIS-7 (A Study to Evaluate the Safety and Anti-cancer Activity of Loncastuximab Tesirine in Combination With Other Anti-cancer Agents in Participants With Relapsed or Refractory B-cell Non-Hodgkin Lymphoma; ClinicalTrials.gov identifer: NCT04970901) is a phase Ib trial evaluating the combination of loncastuximab tesirine with other agents, including gemcitabine, lenalidomide, umbralisib and polatuzumab vedotin for treatment of relapsed B-cell lymphomas.52 Finally, the safety of the addition of loncastuximab tesirine to rituximab plus cyclophosphamide, doxorubicin hydrochloride, vincristine, prednisolone (R-CHOP) is being evaluated in the phase Ib LOTIS-8 trial (A Study to Evaluate the Tolerability, Safety, Pharmacokinetics, and Antitumor Activity of Loncastuximab Tesirine in Combination With Rituximab, Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone (R-CHOP) in Participants With Previously Untreated Diffuse Large B-cell Lymphoma; ClinicalTrials.gov identifier: NCT04974996).53
Conclusions
Decades after the identification of CD19 as a potential therapeutic target in B-cell malignancies, multiple antibody-based therapies targeting CD19 have demonstrated safety and efficacy. Loncastuximab tesirine is the first anti-CD19 ADC to receive regulatory approval. It is also the first approved ADC to deliver a novel class of payload, a PBD dimer. The safety profile of loncastuximab tesirine differs from other approved ADCs and other CD19-targeting agents: it is characterized by the absence of peripheral neuropathy, cytokine release syndrome and immune effector-associated neurotoxicity syndrome; however, it is associated with peripheral oedema, effusions, photosensitivity and elevated GGT. The efficacy against DLBCL appears to be consistent throughout several risk groups. Very limited data suggest loncastuximab tesirine can be used prior to or after CAR T cell therapy,54 but more extensive research needs to validate the sequencing of anti-CD19 agents.
The safety profile, single-agent activity and administration schedule suggest that loncastuximab tesirine could be a valuable addition to combination therapy regimens for B-cell lymphomas. Early data from a single, phase I study suggest that loncastuximab tesirine can be effectively combined with ibrutinib, but ongoing and future studies should continue to explore both additional combinations and indications for this agent.