Bladder cancer is the ninth most common cancer worldwide, with an estimated 430,000 new cases and 165,000 deaths in 2012.1 More than 90% of bladder cancers are of urothelial cell origin and the remaining histological subtypes include either pure forms of histological variants, such as squamous cell carcinoma, adenocarcinoma, small cell carcinoma and sarcoma, or mixed tumours.2 Approximately 30% of patients have muscle-invasive disease at diagnosis, and a proportion present with or develop distant metastases in the course of the disease.3 For over two decades, the prognosis of patients with metastatic or locally advanced, non-resectable bladder cancer has remained poor with platinum-based chemotherapy, with median overall survival (OS) ranging from 9–15 months.4,5 It is clear that better therapeutic options are needed.
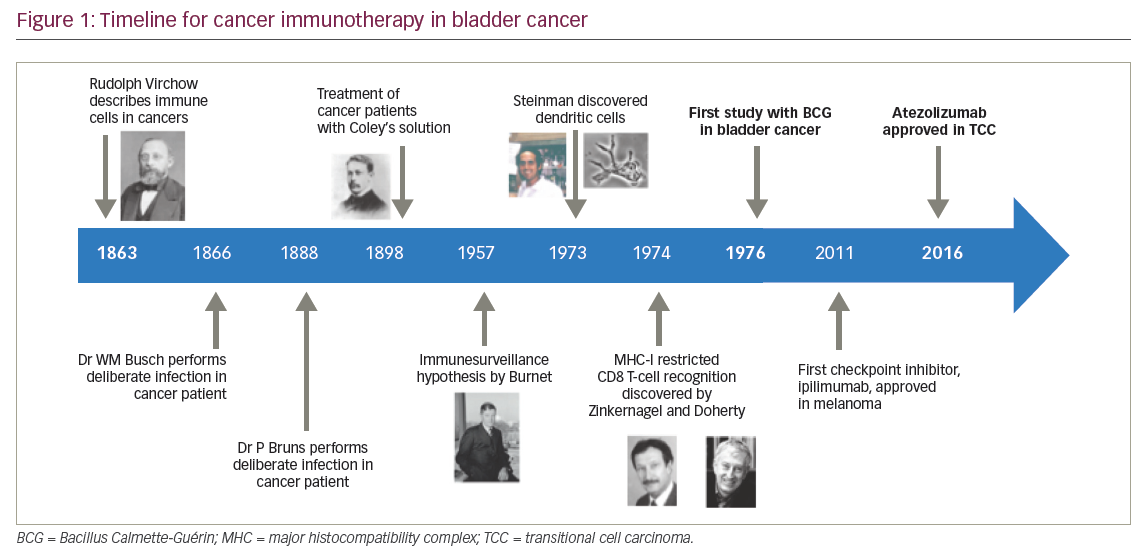
Bladder cancer was the first cancer to be treated with cancer immunotherapy. Figure 1 gives an outline of cancer immunotherapy for bladder cancer.6–10 Intravesical Bacillus Calmette-Guérin (BCG), a form of attenuated mycobacterium, was the first form of immunotherapy used innon-muscle invasive bladder cancer, demonstrating the potential link between immune activation and anti-tumour activity, and gained US Food and Drug Administration (FDA) approval in 1990.11 Since then, the role of immunotherapy has continued to evolve. There is a broad array of immunotherapeutic modalities, including immune checkpoint inhibitors, antineoplastic vaccines, proinflammatory cytokines, chimeric antigen receptors and adoptive T-cell transfer, amongst others. The advent of novel immunotherapy agents, particularly in recent years, has dramatically changed the treatment paradigm for patients with advanced bladder cancer. Multiple immune checkpoint inhibitors have been approved for clinical use in the past 1–2 years, beginning with FDA approval for atezolizumab in May 2016 following progression on platinum-based chemotherapy.10 Here, we will review the current evidence of the clinical usage of immune checkpoint inhibitors with a focus on programmed cell death-1 (PD-1) and programmed death-ligand 1 (PD-L1) inhibitors, as well as their toxicities, potential biomarkers and predictors of response, and provide an outline of future directions in the treatment of patients with metastatic and/or non-surgically curable urothelial bladder cancer.
Currently used therapies in metastatic and non-surgically curable urothelial bladder cancer and unmet clinical needs
Treatment choices in metastatic and/or non-surgically curable bladder cancer are guided by tumour factors (such as histology and disease burden) and patient factors (such as performance status, comorbidities and patient preferences). Cisplatin-based combination chemotherapy is the standard of care in previously untreated patients with advanced urothelial bladder cancer. A phase III trial, in which 405 patients were randomised to either gemcitabine plus cisplatin (GC) or methotrexate, vinblastine, doxorubicin and cisplatin (MVAC), demonstrated similar efficacy (objective response rate [ORR] of 49% versus 46%, time to progression of 7 months, and median OS of 14 months versus 15 months) and less toxicity with GC compared to MVAC.4 Alternative regimens such as dose-dense MVAC12 or triplet chemotherapy with paclitaxel, gemcitabine and cisplatin (PGC)13 have not resulted in significant OS benefit.
Unfortunately, patients with metastatic and/or non-surgically curable bladder cancer who are unable to receive cisplatin, i.e., due to frailty or comorbidities, have an inferior outcome.14,15 The peak incidence of bladder cancer occurs between the ages of 60–70 years old,1 and higher rates of renal insufficiency and poorer performance status are associated with advancing age. For cisplatin-ineligible patients, carboplatin-based regimens are preferred. The EORTC 30986 phase II/III trial randomised 238 chemotherapy-naïve, cisplatin-ineligible patients to gemcitabine plus carboplatin (GCa) versus methotrexate, carboplatin and vinblastine (M-CAVI), demonstrating similar efficacy (ORR 41% versus 30% and median OS 9 versus 8 months) and reduced rate of severe toxicities with GCa compared to M-CAVI.5
Although a significant proportion of patients with metastatic and/or non-surgically curable bladder cancer derive benefit from systemic chemotherapy, responses are short-lived and OS remains poor. Cumulative toxicity also often limits chemotherapy treatment to six cycles with platinum-based treatment.4,5,16 For patients who have relapsed after or progressed on first-line treatment, treatment options are limited and there is no standard second-line chemotherapy. Prognostic factors predictive of shorter survival include low haemoglobin (<10 g/dL), Eastern Cooperative Oncology Group performance status (ECOG PS) ≥1, and the presence of liver metastasis.17 Options include taxanes (such as paclitaxel and docetaxel),18,19 vinflunine and pemetrexed,20 with response rates generally ranging 10–20%. Vinflunine (approved only in the European Union) is the only chemotherapeutic agent studied in a phase III trial,21 where 370 patients were randomised 2:1 to vinflunine or best supportive care (BSC). A statistically significant median OS benefit (6.9 versus 4.3 months) was demonstrated only in the eligible population (n=357) but not the intention-to-treat (ITT) population.21 A meta-analysis by Raggi et al. comparing single- to double-agent second-line chemotherapy demonstrated an improvement in ORR and progression-free survival (PFS), but not median OS with doublet chemotherapy.22
Given the poor prognosis of patients with advanced bladder cancer, there is a large unmet medical need for this group of patients, particularly for those with progressive disease following first-line therapy, as well as the significant proportion of patients who are cisplatin-ineligible. The first immune checkpoint inhibitor, ipilimumab, an anti-cytotoxic T-lymphocyte antigen 4 (CTLA4) antibody, gained FDA approval for use in advanced melanoma in 201123 and since then cancer immunotherapy has revolutionised the field of oncology, with ever-expanding use in cancer treatment, including in patients with advanced urothelial bladder cancer. In a subset of patients, promising evidence points to durable responses and improved long-term survival.28–31,33–38,40 Five immune checkpoint inhibitors (pembrolizumab, nivolumab, atezolizumab, durvalumab and avelumab) targeting the PD-1 or its ligand PD-L1 have been approved by the FDA for use in patients with advanced bladder cancer.
Overview of immunotherapy agents and their mode of action
The immune system, which can be divided into the innate and adaptive components, each with their distinct effector cells and downstream actions, plays a critical role not just in protection against infectious pathogens, but also in tissue homeostasis and the elimination of damaged or malignant cells. The immune system itself is tightly modulated to prevent aberrant activation, and to allow an optimum response. One of the many complex mechanisms of auto-regulation is through T-cell coinhibitory signalling, which counter balances costimulatory signalling following antigen presentation. When engaged by their ligands, the T-cell surface proteins PD-1 and CTLA-4 result in inhibition of downstream pathways that normally lead to increased T-cell activity and proliferation.
In a 2011 update, Hanahan and Weinberg alluded to immune avoidance as one of the emerging hallmarks of cancer, in addition to the original six hallmarks first presented in the year 2000.24,25 Tumours in general (including bladder cancer) can suppress anti-tumour activity through the increased expression of inhibitory signals, for example via the upregulation of PD-1 ligands (e.g. PD-L1) through transcriptional or post-transcriptional mechanisms, thus escaping T-cell mediated tumour activity.26,27 Therapeutic immune checkpoint inhibitors are therapies that “release the break” on the immune system – they do not target cancer cells directly, but instead target lymphocyte receptors or their ligands to enhance endogenous anti-tumour activity.
Immunotherapy for advanced urothelial bladder cancer in the second-line or later setting
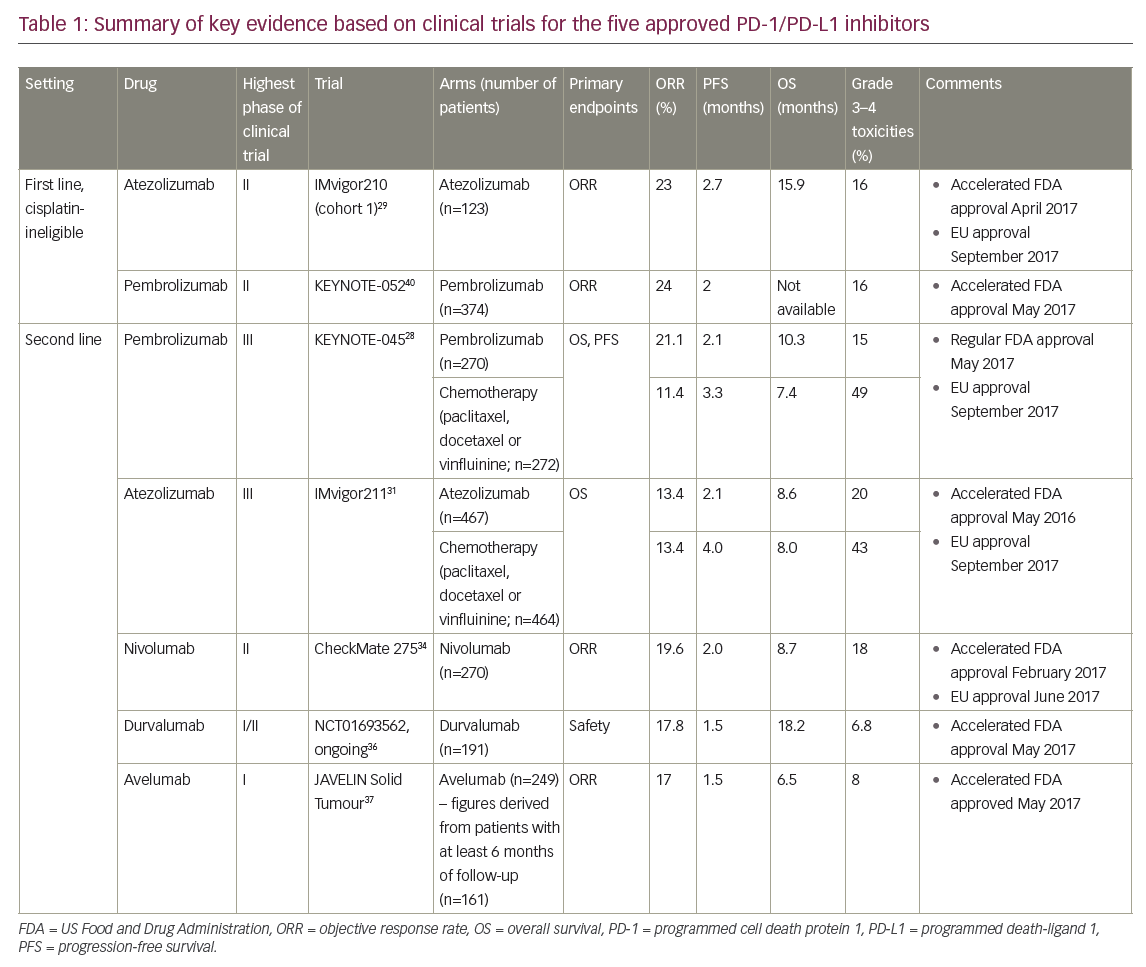
Of the PD-1 inhibitors (pembrolizumab and nivolumab) and PD-L1 inhibitors (atezolizumab, durvalumab and avelumab), only two agents, atezolizumab and pembrolizumab, are approved in the first-line setting for cisplatin-ineligible patients, whereas all five are approved in previously treated patients following progression on platinum-based chemotherapy. Of these, data from randomised phase III trials exist only for pembrolizumab and atezolizumab,28,31 and thus far a proven OS benefit has been established only with pembrolizumab. Pembrolizumab is the only agent with regular FDA approval, with the other agents having accelerated approval based on phase II data29–30,33–36,38,40 in the context of historical control data, thus highlighting the need for effective treatments in this setting. Key findings are summarised in Table 1.
Pembrolizumab
Pembrolizumab is an anti-PD-1 inhibitor that has been shown to prolong survival in patients with advanced urothelial carcinoma who have progressed after platinum-based chemotherapy for advanced disease or have recurrence within 12 months following platinum-based adjuvant or neoadjuvant therapy for localised muscle-invasive disease.28 In the practice-changing, open-label, randomised phase III KEYNOTE-045 trial, 542 patients were randomised 1:1 to receive pembrolizumab 200 mg every 3 weeks or the investigator’s choice of chemotherapy with paclitaxel, docetaxel or vinflunine until Response Evaluation Criteria in Solid Tumours version 1.1 (RECIST v1.1)-defined disease progression, development of an unacceptable level of toxic effects or the completion of 2 years of pembrolizumab therapy.28 Cross-over was not permitted. Patients were enrolled regardless of the level of PD-L1 expression. Thirty percent of these patients had tumour PD-L1 combined positive score (defined as the percentage of PD-L1-expressing tumour and infiltrating immune cells [IC] relative to the total number of tumour cells) of ≥10%, and 55% had PD-L1 combined positive score of <1% (as assessed based on the Dako 22C3 assay [Agilent, Santa Clara, CA, US]). The co-primary endpoints were OS and PFS.
Pembrolizumab was associated with significantly longer OS independent of PD-L1 expression and was well tolerated with a lower rate of treatment-related adverse events compared to chemotherapy. The median OS was improved in the total population (10.3 months in the pembrolizumab group versus 7.4 months in the chemotherapy group, hazard ratio [HR] 0.73; 95% confidence interval [CI] 0.59–0.91, p=0.002) and in patients who had a PD-L1 combined positive score of ≥10% (8.0 months in the pembrolizumab group and 5.2 months in the chemotherapy group: HR 0.57; 95% CI 0.37–0.88, p=0.005). The response rate was higher with pembrolizumab compared with chemotherapy (21% versus 11%). The median duration of response was not reached in the pembrolizumab group and was 4.3 months in the chemotherapy group. There was no significant difference in PFS in the total population (2.1 months in the pembrolizumab group versus 3.3 months in the chemotherapy group) or among patients who had a tumour PD-L1 expression of ≥10% (HR 0.89, 95% CI 0.61–1.28, p=0.24). The 12-month OS rates were 44.0% versus 31.0%, and the 18-month OS rates were 36.0% and 20.5% respectively.28
The OS benefit with pembrolizumab was similar regardless of investigator’s choice of chemotherapy, and the median OS of 7.4 months observed with chemotherapy is consistent with historical data for second-line chemotherapy treatment.22 ORR and OS benefit was seen regardless of PD-L1 expression, although patients with higher levels of PD-L1 expression appear to benefit more, e.g. HR for death for those with tumour PD-L1 combined positive score of <1% was 0.89 (0.66–1.20), 0.61 (0.43–0.86) for those with a PD-L1 combined positive score of ≥1%, 0.80 (0.61–1.05) for those with a PD-L1 combined positive score of <10%, and 0.57 (0.37–0.88) for those with a PD-L1 combined positive score of ≥10%.28
Rates of grade 3–5 toxicities were lower in the pembrolizumab group compared to the chemotherapy group (15% versus 49%), as was treatment-related discontinuation of therapy (5.6% versus 11.0%). The most common treatment-related adverse events of any grade were pruritus (9.5%), fatigue (13.9%) and nausea (10.9%) in the pembrolizumab group, and alopecia (37.6%), fatigue (27.8%) and anaemia (24.7%) in the chemotherapy group. The only immune-related adverse events of grade 3 and above severity that were observed in two or more patients who were treated with pembrolizumab were pneumonitis (2.3%), colitis (1.1%) and nephritis (0.8%). There was one treatment-related death, which was due to pneumonitis.28
Atezolizumab
The initial accelerated FDA approval in May 2016 for atezolizumab, an anti-PD-L1 inhibitor, for the treatment of advanced urothelial carcinoma following progression on platinum-based chemotherapy, was based on single-arm phase II data that demonstrated safety and activity of the drug.29 PD-L1 expression status was assessed using the companion Ventana SP142 assay (Ventana Medical Systems Inc., Tucson, AZ, US), and was defined by the percentage of PD-L1-positive IC in the tumour microenvironment: IC0 (<1%), IC1 (≥1% but <5%), and IC2/3 (≥5%). Cohort two of the IMvigor 210 trial (n=310) showed that atezolizumab resulted in an ORR of 15% for all patients, 26% in those with ≥5% PD-L1 expression and 8% for those with <1% PD-L1 expression. The median OS was 11.4 months (95% CI 9.0–not estimable) in patients in the IC2/3 group, 8.8 months in the IC1/2/3 group and 7.9 months for the entire cohort of patients. Durable responses with long-term clinical benefit were seen, with the median duration of response not reached after 17.5 months of follow-up.30
Phase III data came from the IMvigor 211 trial, in which 931 patients with metastatic urothelial carcinoma and progression after platinum-based chemotherapy were randomised 1:1 to atezolizumab (1,200 mg every 3 weeks) or to investigator’s choice of chemotherapy (paclitaxel, docetaxel or vinfluinine).31 No prespecified crossover was planned per protocol. The primary endpoint of OS was tested hierarchically in prespecified populations: IC2/3, followed by IC1/2/3, followed by the ITT population. Median follow up was 17.3 months.
Unexpectedly, the primary endpoint of OS improvement with atezolizumab was not met in patients with metastatic urothelial carcinoma with at least 5% PD-L1 expression on tumour-infiltrating IC (who comprised 25% of the total study population), although the safety profile for atezolizumab was more favourable. Median OS for the atezolizumab group was 11.1 months (95% CI 8.6–15.5), versus 10.6 months for the chemotherapy group (95% CI 8.4–12.2), with a stratified HR of 0.87 (95% CI 0.63–1.21, p=0.4).The lack of a statistically significant OS benefit with atezolizumab compared to chemotherapy precluded further formal statistical comparisons and rendered subsequent analyses exploratory in nature. In the ITT population, median OS was 8.6 months with atezolizumab and 8.0 months with chemotherapy, and the 12-month OS rate was 39.2% with atezolizumab (95% CI 34.8–43.7) and 32.4% with chemotherapy (95% CI 28.0–36.8).31
In the IC2/3 group, ORR was 23% with atezolizumab and 21.6% with chemotherapy, and in the ITT population, response rates were 13.4% in both treatment arms. In the IC2/3 group, median duration of response was 15.9 months with atezolizumab (95% CI 10.4–not reached) and8.3 months with chemotherapy (95% CI 5.6–13.2); in the ITT population, median duration of response was 21.7 months with atezolizumab(95% CI 13.0–21.7) and 7.4 months with chemotherapy (95% CI 6.1–10.3).31
In this study, tumour mutation burden was analysed using the FoundationOne® assay (Foundation Medicine Inc., Cambridge, MA, US),32 and was categorised as high (at or above the median) or low (less than the median). In an exploratory biomarker analysis, patients with high tumour mutation burden had longer median OS for those treated with atezolizumab than for those treated with chemotherapy (11.3 months versus 8.3 months; HR 0.68, 95% CI 0.51–0.90), whereas for those with low tumour mutation burden, survival was similar between groups (8.3 months versus 8.1 months; HR 1.00, 95% CI 0.75–1.32). In patients with high tumour mutation burden and PD-L1 IC2/3 samples (n=96), median survival for patients given atezolizumab was 17.8 months(95% CI 9.7–not estimable) versus 10.6 months (95% CI 8.2–14.3) for those given chemotherapy (HR 0.50, 95% CI 0.29–0.86). In the ITT population, grade 3–4 treatment-related adverse events were less common with atezolizumab than with chemotherapy (20% versus 43%), as were rates of treatment discontinuation due to adverse events (7% versus 18%).31
The reasons for a lack of OS benefit with atezolizumab remain unclear, especially given the promising data from previous phase I and II studies.29,30 Several possible explanations exist. Firstly, theKaplan-Meier survival curves separation occurred relatively late at about 11 months, compared to 4 months in KEYNOTE-045. Secondly, PD-L1 expression also appeared to correlate with improved response rates both to atezolizumab and chemotherapy in this study.31 In addition, unlike in KEYNOTE-045 where there was a relatively equal proportion of patients receiving one of the three chemotherapy options, in IMvigor 211, the majority of patients received vinflunine (55%), as compared to paclitaxel (33%) and docetaxel (12%). Although previous data have suggested similar OS rates with vinflunine and taxanes in the second-line setting,22 to date no randomised controlled trials have been conducted comparing these agents directly. Hence, the possibility of the effect of a mixed control group on OS outcomes cannot be excluded. While there was no OS benefit demonstrated with IMvigor 211, the duration of response and 12-month survival rates were improved with atezolizumab compared to chemotherapy, and treatment was better tolerated.31
Nivolumab
Nivolumab is an anti-PD-1 antibody that is approved by the US FDA for the treatment of advanced urothelial cancer following progression on platinum-based chemotherapy based on phase I and II studies demonstrating significant activity of the drug in this group of patients.33,34
In the larger of the published phase II studies, the CheckMate 275 trial, 270 patients received nivolumab 3 mg/kg every 2 weeks and the primary endpoint was overall objective response.34 PD-L1 expression was assessed using the Dako 28–8 assay. Fifty-three percent of patients had PD-L1 expression of <1% and 30% of patients had PD-L1 expression of ≥5%. The median duration of follow up was 7 months. The ORR was 19.6% in the total population, 28.4% in those with PD-L1 expression of ≥5%, 23.8% in those with PD-L1 expression of ≥1% and 16.1% in those with PD-L1 expression of <1%. At 7-month follow up, OS for the entire cohort was 8.7 months; for those with PD-L1 expression of <1 and ≥1%, median OS durations were 6.0 and 11.3 months, respectively. The median duration of response was not reached. Grade 3–4 adverse events occurred in 18% of patients, and there were three treatment-related deaths (pneumonitis, acute respiratory failure and cardiovascular failure).
Durvalumab
Durvalumab is an anti-PD-L1 antibody that was granted accelerated FDA approval for the treatment of patients with advanced urothelial cancer based on an ongoing phase I/II study.35,36 Durvalumab was administered at 10 mg/kg every 2 weeks for up to 12 months in 191 patients with metastatic urothelial carcinoma, and the primary endpoint was safety. ORR with durvalumab was 17.8%, median PFS was 1.5 months and median OS was 18.2 months. The response rate was higher in high PD-L1 expression tumours (defined as PD-L1 expression of ≥25% on tumour or IC on the Ventana SP262 assay) compared with low or negative PD-L1 expression (28% versus 5%). At a median follow up of 5.8 months, the median duration of response had not been reached. The 1-year OS rate was 55%. Grade 3–4 treatment-related and immune-mediated adverse events occurred in 6.8% and 2.1% of patients respectively, and treatment-related adverse events led to discontinuation of three patients (1.6%), two of whom had immune-mediated adverse events that led to death (autoimmune hepatitis and pneumonitis).35,36
Avelumab
Avelumab, an anti-PD-L1 antibody, was approved in the treatment of metastatic urothelial carcinoma after platinum failure based on a single arm phase I trial. In a pooled analysis of two phase I expansion cohorts, 249 PD-L1 unselected patients received avelumab 10 mg/kg every 2 weeks.37 The primary endpoint was best overall response. PD-L1 status was based on numbers of tumour cells with plasma membrane PD-L1 expression at any intensity using a staining cut off of 5% or higher on the Dako 73-10 assay. Of the 83% of patients with evaluable samples, 82 (33%) had PD-L1-positive tumours and 124 (50%) had PD-L1-negative tumours.
In patients with at least 6 months of follow up (n=161), the ORR was 17%, with higher response rates in patients with PD-L1 positive tumours compared with those with PD-L1 negative tumours (24% versus 14%). Median OS was 6.5 months. Responses were durable; the median duration of response was not reached and the estimated proportion of responses lasting at least 24 weeks was 96% (95% CI 77–99). Grade 3 or worse treatment-related adverse events occurred in 8% of patients and there was one treatment-related death (pneumonitis).
Immunotherapy for advanced bladder cancer in the first-line setting for cisplatin-ineligible patients
Atezolizumab
Atezolizumab was granted accelerated approval by the FDA in April 2017 as first-line treatment for patients with locally advanced and metastatic urothelial carcinoma who are cisplatin-eligible. Supporting evidence came from cohort 1 of the phase II IMvigor 210 trial.38 Cisplatin-ineligibility criteria include glomerular filtration rate of >30 mL/min and <60 mL/min (Cockcroft-Gault formula), grade 2 or higher hearing loss or peripheral neuropathy, or an ECOG PS of 2. Neoadjuvant or adjuvant chemotherapy or radiation was permitted if more than 12 months had elapsed between treatment and recurrence. One hundred and twenty-three patients were enrolled to receive atezolizumab at 1,200 mg every 3 weeks, of whom 119 received one or more doses of atezolizumab. The primary endpoint was ORR, assessed in a hierarchical fixed-sequence manner for three pre-specified subgroups (PD-L1 IC2/3, followed by IC1/2/3 and followed by all patients, versus a control ORR of 10%).38
Thirty-three percent of patients had IC0, 40% had IC1 and 27% had IC2/3, a distribution similar to previous study populations.29 The majority of patients were cisplatin-ineligible due to renal impairment (70%) and ECOG PS 2 (20%), and 7% of patients had both renal impairment and ECOG PS 2; 14% had hearing loss of ≥25 dB and 6% had grade 2 or higher peripheral neuropathy. Twenty-one percent of patients were aged 80 years and above.38
At a median follow up of 17.2 months, ORR was 23% in all patients(95% CI 16–31) and responses occurred across all PD-L1 subgroups. ORR was 28% (95% CI 14–47) in the IC2/3 subgroup, 21% (95% CI 10–35) in the IC1 subgroup, and 21% (95% CI 9–36) in the IC0 subgroup. Median OS was 15.9 months in all patients (95% CI 10.4–not estimable), 12.3 months in IC2/3 patients, and 19.1 months in IC0/1 patients. Median PFS was2.7 months. Median duration of response was not reached.38 OS surpassed historical rates for cisplatin-ineligible patients, which was in the range of 8–9 months for patients receiving GCa or M-CAVI.5
Exploratory biomarker assessments revealed that responses were more frequent in patients with the Cancer Genome Atlas (TCGA) luminal II subtype and higher mutation load. Interestingly, amongst the 85 patients (71%) who had primary bladder or urethral tumours, ORR was 17%(95% CI 9–26), whereas amongst the 33 patients (28%) with upper tract tumours, a group historically associated with a poorer prognosis,39 ORR was higher at 39% (95% CI 23–58). To investigate a possible basis for improved outcomes in these patients, baseline covariates including tumour mutation load, T-effector gene expression, TCGA subtype and baseline tumour burden were assessed. However, no significant differences were found in these factors between patients withupper-tract and lower-tract primary tumours.38
Grade 3 or 4 treatment-related adverse events occurred in 16% of patients and there was one treatment-related death. Eight percent of patients had an event leading to treatment withdrawal. There were no major safety differences across PD-L1 subgroups. Post-protocol treatment, defined as any treatment administered after progression on atezolizumab before study discontinuation, was reported for 25 patients (21% of the total cohort), of whom the majority (14) received carboplatin-gemcitabine.38
Pembrolizumab
Evidence for the use of pembrolizumab in the first-line setting comes from the ongoing single arm, phase II KEYNOTE-052 trial.40 Three hundred and seventy-four cisplatin-ineligible patients with locally advanced and unresectable or metastatic urothelial cancer were enrolled to receive pembrolizumab at 200 mg every 3 weeks, and 370 patients received at least one dose of pembrolizumab. PD-L1 staining was scored based on the percentage of cells (tumour cells, macrophages or lymphocytes) expressing PD-L1 in a tumour biopsy. Two combined positive score cut-offs were used: a 1% cut-off and a strongly positive (high) PD-L1 expression cut-off. The primary endpoint was ORR. Secondary objectives included establishment of a cut-off level for PD-L1 strongly positive expression status.40
Reasons for cisplatin-ineligibility included renal dysfunction (49%), ECOG PS 2 (32%), both renal dysfunction and ECOG PS 2 (9%), and other reasons (9%). Twenty-nine percent of patients were aged 80 years and above. Seventy-five percent of patients had a PD-L1 expression of at least 1%, and 30% of patients had a PD-L1 expression of at least 10%. The median follow-up duration for the current analysis was relatively short at 5 months.40
The ORR was 24%. Responses were seen across all categories of PD-L1 expression, although higher response rates were seen in those with a higher combined PD-L1 score of 10% (ORR 38%, 95% CI 29–48), which was determined to be the optimum high cut-off value. Responses to pembrolizumab were also seen irrespective of reason forcisplatin-ineligibility, comorbidities or age. Thirteen out of 59 patients (22%) with a primary upper tract tumour had an objective response, as did 70 out of 247 patients (28%) with a primary lower tract tumour. As of data cut-off, the median duration of response was not reached(95% CI 9 months–not reached). One hundred and thirty-seven patients (37%) were still receiving treatment and 74 responses (83%) were ongoing, with 78% of responses lasting at least 6 months. Sixteen percent of the patients had a grade 3 or worse treatment-related adverse event, and there was one treatment-related death. Nineteen patients (5%) discontinued treatment because of adverse events.40
There were some differences between the study populations in IMvigor 21038 and KEYNOTE-052.40 The study population in KEYNOTE-052 was substantially larger compared to IMvigor 210 (374 versus 123), although follow-up duration was shorter (5 versus 17 months). The patients in KEYNOTE-052 were slightly older (≥80 years: 29% versus 21%), had more advanced disease (visceral disease: 85% versus 66%) and poorer performance status (ECOG PS 2: 41% versus 27%). A larger proportion had lower tract tumours – 81% of patients in the KEYNOTE-052 study had lower tract tumours compared to 71% in the IMvigor 210 population – and response rates were slightly better in patients with lower tract tumours in KEYNOTE-052 whereas the reverse was seen in IMvigor 210. Despite these differences, the ORRs were similar between the two studies. Of note, OS data for KEYNOTE-052 is not yet published due to the short follow-up time in the current analysis.
Toxicities
Because immune checkpoint inhibition plays an important role in the maintenance of self tolerance, immune checkpoint blockade can result in unchecked systemic immune responses that may affect virtually any organ system of the body, with a spectrum of toxicities distinct from that of cytotoxic chemotherapy. As examples, immune-related adverse events may result in mucocutaneous, pulmonary, gastrointestinal, renal, endocrine (e.g. thyroid, adrenal, pituitary), cardiovascular, musculoskeletal and neurological toxicities. Various clinical trials28-31,33–38,40 have generally excluded patients with autoimmune disease (e.g. depending on activity and the use of systemic immunosuppressive treatment). While the rates of grade 3 and above toxicities are lower compared to chemotherapy, severe toxicities and treatment-related death can occur. The presentation of immune-related adverse events may be non-specific, and a high level of clinical suspicion may be required. Patients may benefit from multidisciplinary care.
In general, the safety profiles shown in the trials presented above demonstrate similarity in the setting of bladder cancer compared to other tumour types with the use of PD-1/PD-L1 checkpoint inhibitors. In addition, no unexpected toxicities were seen in the two first-line phase II trials (IMvigor 21038 and KEYNOTE-05240) despite the older population and lower ECOG PS of patients in these studies. The American Society for Clinical Oncology (ASCO) has recently published a comprehensive guideline for the management of immunotherapy-related toxicities categorised by organ system and grade.41
Biomarkers and predictors of response
The totality of current available evidence suggests that PD-L1 expression may be a useful biomarker for selecting patients who are more likely to benefit from treatment with PD-1/PD-L1 inhibitors.42 However, selection based on PD-1 expression does not always enrich for responders, and clinical benefit is seen in PD-L1 negative patients as well. In addition, durable response is seen only in a fraction of patients. The use of PD-L1 expression as a biomarker may be affected by several pre-analytical and analytical factors, such as tumour spatial heterogeneity, measurements on tumour cells and/or tumour-infiltration lymphocytes or other ICs in the tumour microenvironment, dynamic expression over time (e.g. in response to platinum or other chemotherapy), a lack of standardised test assays for defining PD-L1 positivity (with different sensitivities and specificities amongst the different assays), as well as the different thresholds for positivity used in tumour types and clinical trials, including those outlined above. This has resulted in inconsistencies in the literature and uncertainties regarding how best to select patients who will benefit most from PD-1/PD-L1 inhibitors.43 Different patients may have discordant PD-L1 results depending on the assay used and the timing and type of tissue used for testing (e.g. fresh or archival). In addition, the immense complexity of immune regulation through mechanisms elucidated, and as yet unknown, also contributes to the inconsistencies in the predictive value of PD-L1 expression.
Beyond PD-L1, other biomarkers under examination include tumour mutational burden, mismatch repair status and TCGA molecular subtypes. It has been observed that increased mutational, and hence theoretically, neoantigen burden in tumours is associated with improved efficacy and durable clinical benefit in patients treated with immune checkpoint inhibitors. In fact, urothelial carcinoma has the third highest mutation burden across tumour types after lung cancer and melanoma.44 Data from IMvigor 210 suggests an association between higher tumour mutation burden and response to atezolizumab.29,38 Mutation load was also associated with OS; patients with the highest mutation load(quartile 4) had significantly longer survival compared with patients in quartiles 1–3.38 TCGA subtype also appears to predict response to immune checkpoint blockade. In cohort 2 of IMvigor 210, objective responses to atezolizumab were seen in all TCGA subtypes but were significantly higher in the luminal cluster II subtype (ORR 34%, p=0.0017) compared with luminal cluster I (10%) and basal clusters III and IV(16% and 20% respectively).29 However, the sample sizes are small and further studies are needed to validate these findings. Patients with higher mutation burden also often have mismatch repair deficiencies, and mismatch repair status has also been shown to predict clinical benefit of immune checkpoint blockade.45 In addition, the presence of a large and functional T-cell population is needed in order to achieve successful anti-tumour effect with immune checkpoint inhibition.
Other biomarkers under study with potential predictive value include gene expression profiles (such as interferon gamma expression), tumour-infiltrating cytotoxic T-lymphocytes and PD-L2 (another ligand capable of binding to PD-1).43 In future, standardisation of PD-L1 expression or use of a composite score encompassing multiple biomarkers may better identify patients who will benefit most from immune checkpoint inhibition and thus guide patient selection. Nevertheless, it should be noted that current approval for the five PD-1/PD-L1 inhibitors for use in urothelial bladder cancer is independent of PD-L1 expression whether in the first- or second-line setting.
Other novel agents
Despite the promise of immune checkpoint inhibitors, less than half of treated patients have an objective response to these agents. In addition, the ability of cancer cells to continuously adapt to their microenvironment and to therapeutic agents through genomic instability and other acquired resistance mechanisms eventually leads to treatment failure and disease progression in the majority of patients. Treatment strategies incorporating new agents to target mechanisms of de novo or acquired resistance to immune checkpoint inhibition (such as interferon gamma signalling) may prove useful.46,47 Further research is needed in this area.
Apart from PD-1/PD-L1 checkpoint inhibitors, other agents are being studied. For example, ramucirumab, an anti-vascular endothelial growth factor receptor 2 antibody has been studied in the phase III RANGE trial for patients with advanced urothelial carcinoma who have progressed following platinum-based chemotherapy.48 Five hundred and thirty patients (of whom 64% had primary bladder tumours) were randomly assigned to docetaxel plus ramucirumab or docetaxel alone. Eighteen patients in the combination arm and 26 patients in the control arm had previously received immune checkpoint inhibitor therapy. The ORR was increased with combination therapy (25.4% versus 14%) and median PFS was increased as well (4.1 versus 2.8 months; HR 0.76, 95% CI 0.61–0.94, p=0.012). Of the patients who previously received an immune checkpoint inhibitor, five of 14 (36%) who were allocated ramucirumab and two of 19 (11%) who were assigned placebo achieved an objective response to treatment. The safety profile was generally similar between the two arms. Rates of grade 3 and above haemorrhage (including haematuria) were similar at 3% in the combination arm and 5% in the control arm. There were four sepsis-related deaths (2%) and one neutropaenic-sepsis-related death in the ramucirumab arm and none in the control arm. OS data remains immature.48 Ramucirumab is not currently approved for this indication. Besides chemotherapy, ramucirumab in combination with PD-1 or PD-L1 inhibitors has shown promising clinical activity in multiple tumour types including urothelial carcinoma in the phase I setting and further studies are ongoing.49
Molecular analysis has also identified genetic alterations in urothelial carcinoma that may be targeted by drugs already approved in other settings or tumour types. These include mutations in pathways that mediate cell growth, survival and angiogenesis, such as phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) and ERB2.44,50 For example, activating PI3KCA mutations are found in about 20% of urothelial carcinomas44 and may potentially be targeted by the mTOR inhibitor everolimus, as used in breast cancer,51 and the PI3K inhibitor idelalisib, as used in non-Hodgkin’s B cell lymphoma and relapsed chronic lymphocytic leukaemia.52,53 ERBB2 amplifications and somatic mutations have been described in urothelial bladder carcinomas with a frequency of about 12%,44 thus potentially rendering ERB2-mutated bladder cancers susceptible to anti-human epidermal growth factor receptor 2 (HER2) targeted therapies such as trastuzumab, pertuzumab and lapatinib.54 The increasing use of next-generation sequencing in clinical practice, together with the increasing number of therapeutic options available for targetable genomic alterations, is likely to result in a trend toward a targeted therapeutic strategy for patients across various tumour types including urothelial carcinomas. However, more prospective evidence is needed.
Future directions and unanswered questions
Despite the obvious advances made, many questions remain to be addressed. Most of the trials presented above have reported relatively short durations of PFS despite improvements in ORR. Indeed, immunotherapy has been shown to demonstrate delayed and atypical response kinetics such as the phenomenon of pseudoprogression (temporary enlargement of lesions due to immune cell infiltration).55 The issue of optimising efficacy evaluation in order to reduce the risk of premature cessation of treatment in patients who may benefit from immunotherapy remains to be addressed. Immune-related RECIST criteria (irRC) has been developed, incorporating updated definitions to better assess the efficacy with immunotherapy which might otherwise be underestimated with conventional RECIST 1.1 criteria.55 Prospective evaluations of irRC and RECIST 1.1 are needed.
Secondly, no head-to-head comparisons have been carried out amongst the various PD-1/PD-L1 inhibitors in the setting of urothelial cancer thus far, and there is a lack of direct evidence to suggest a preferred checkpoint inhibitor over another. Currently, pembrolizumab in the setting of progression following platinum-based chemotherapy is the only agent with proven efficacy in a randomised phase III trial, and which has gained regular FDA approval. Although all five drugs appear to have similar efficacy and safety profiles, cross-trial comparisons have their inherent limitations. The failure to demonstrate OS benefit withsecond-line atezolizumab in the phase III IMvigor 211 trial alsohighlights the need for caution in the use of drugs approved based on phase I or II trials.31
Further on, we do not know if treatment with a different immune checkpoint inhibitor will lead to a meaningful response following prior immune checkpoint inhibitor failure (e.g. if there will be benefit with treatment with a PD-L1 inhibitor following progression after a PD-1 inhibitor).
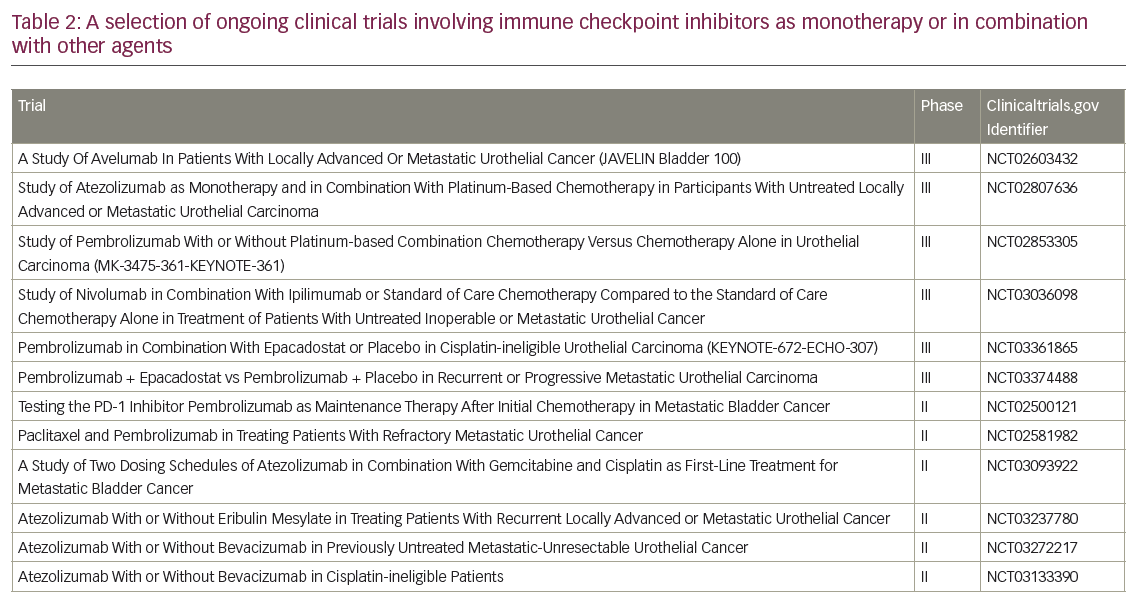
Next, is there an ideal duration of therapy to prolong tumour response and disease control? How about the role of maintenance treatment in patients who have not progressed following platinum-chemotherapy or immunotherapy? Finally, is there an optimum sequential or combination approach with chemotherapy and targeted therapy, especially in the trend toward next-generation sequencing and precision medicine? Many clinical trials are underway involving immunotherapy beyond checkpoint inhibitors, such as phase I/II adoptive T-cell therapy studies,56 as well as checkpoint inhibitors alone or in combination with other therapies (such as anti-CTLA4 and indoleamine 2,3 dioxygenase [IDO] inhibitors) in the first- and subsequent-line therapies of advanced urothelial cancer. Selected ongoing trials are presented in Table 2.
Conclusion
Immune checkpoint inhibition has changed the standard of care in the treatment of metastatic and/or non-surgically curable urothelial cancer where little progress has been made over the past 20 years. Evidence discussed above has demonstrated proven clinical benefit and acceptable safety profile even in elderly patients with pre-existing comorbidities. First-line response rates are modest compared to platinum-based chemotherapy, although durable benefit is seen in a proportion of patients, which is extremely rare with chemotherapy in metastatic and/or non-surgically curable bladder cancers. These modest response rates of immune checkpoint inhibitors may be due the paucity of a functioning T-cell population. Clearly, more data is needed to define biomarkers and parameters that will select for patients who will derive the greatest benefit with immune checkpoint inhibitors or immunotherapy in the broader sense. The optimum sequence and/or combination of systemic treatments including immunotherapy, chemotherapy and targeted therapy remains to be defined.