The majority (60–75%) of all breast cancers have oestrogen and/or progesterone receptors.1
Endocrine treatment constitutes the therapeutic backbone for patients with this cancer subtype
unless there is a visceral crisis or concern/proof of endocrine resistance,2 as recommended by
the third European School of Oncology (ESO)/European Society for Medical Oncology (ESMO)
international consensus guidelines for Advanced Breast Cancer (ABC 3)3 and the National
Comprehensive Cancer Network (NCCN) guidelines.4 Current endocrine therapy includes: selective
oestrogen receptor modulators, aromatase inhibitors, and selective oestrogen-receptor degraders
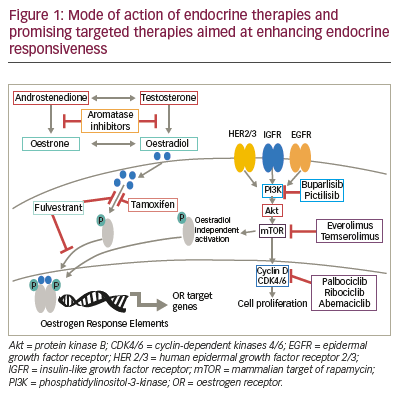
(Table 1), and the modes of action of these therapies are outlined in Figure 1. Not all patients have
a response to first-line endocrine therapy (primary or de novo resistance). Such resistance occurs
in approximately 40% of patients with hormone receptor (HR)-positive breast cancer, and even
patients who do respond eventually exhibit acquired resistance.5 Cytotoxic chemotherapy is also
considered a first-line treatment option in patients diagnosed with HR-positive breast cancer. The
decision for chemotherapy or endocrine therapy depends on a number of factors, outlined below,
and there is a wide variation in the use of these treatments.6
Endocrine resistance
Several molecular mechanisms have been proposed to underlie endocrine resistance, including:
loss of oestrogen receptor expression; altered activity of oestrogen-receptor co-regulators;
deregulation of apoptosis and cell cycle signalling; hyperactive receptor tyrosine kinase; and
stress/cell kinase pathways.7 The oestrogen receptor may be activated in a ligand-independent
manner via intracellular signal transduction pathways mediated either by the phosphatidylinositol-
3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway,
(Figure 1) or the mitogen-activated protein kinase (MAPK) pathway which promotes oestrogen
receptor phosphorylation and subsequently, activation.8,9 In addition, mutations in the ESR1
gene have recently attracted attention as an important mechanism for endocrine resistance
in metastatic breast cancer (MBC). These mutations occur in approximately 20–40% of patients
with metastatic oestrogen receptor-positive disease who received endocrine therapies, with the
higher occurrence in more advanced patients.10 Clustered in a ‘hotspot’ within the ligand-binding
domain (LBD) of the oestrogen receptor, these mutations lead to ligand-independent oestrogen
receptor activity that promotes tumour growth, and partial resistance to endocrine therapy, and
potentially enhanced metastatic capacity.10 The purpose of this article is to provide a concise
overview of endocrine therapeutic strategies for MBC, including studies with cohorts in first-line
therapy, second-line and beyond.
Endocrine therapy
Tamoxifen
Tamoxifen, first described in the treatment of advanced breast cancer
in 1971,11 is the oldest selective oestrogen receptor modulator in clinical
use. In the 1990s, tamoxifen became standard first-line treatment based
on randomised, controlled trials, demonstrating comparable efficacy to
megestrol acetate or aminoglutethimide, but with superior tolerability.
Subsequently, tamoxifen was replaced by third-generation aromatase
inhibitors (letrozole, anastrozole, exemestane), which have demonstrated
3–4 months improvement in progression-free survival (PFS) in a range of
randomised, controlled trials, for example, in postmenopausal women
with oestrogen synthesis occurring mainly in peripheral tissues, but do
not benefit in overall survival (OS) (Table 1).12-16
Fulvestrant
Fulvestrant is a selective oestrogen receptor degrader that blocks
oestrogen receptor dimerisation and DNA binding, inhibiting nuclear
translocation while increasing turnover of the oestrogen receptor
(Figure 1). This leads to inhibition of oestrogen signalling via a reduction
of oestrogen receptor expression and accelerated oestrogen receptor
degradation.17 A multicentre, double-blind, randomised trial, in patients
with metastatic/locally advanced breast cancer comparing treatment
with fulvestrant (250 mg/month) versus tamoxifen (20 mg/day) found no
significant difference between fulvestrant and tamoxifen for the primary
end point of time to progression (TTP).18 Similarly, in a double-blind,
randomised trial comparing the efficacy and tolerability of fulvestrant
versus anastrozole in postmenopausal women with advanced breast
cancer progressing on prior endocrine therapy, fulvestrant was found to
be at least as effective as anastrozole, with efficacy endpoints slightly
favouring fulvestrant.19
Initial investigation of fulvestrant in breast cancer used a dose of 250 mg,
which the latest evidence suggests is suboptimal. Whereas fulvestrant
250 mg is sufficient to competitively inhibit binding of oestradiol to
the oestrogen receptor, oestrogen receptor downregulation is a dosedependent
process.20 At this dose, inhibition of oestrogen receptor
transcription may occur but with incomplete oestrogen receptor
degradation, i.e., so that both mechanisms of action of fulvestrant are
not being utilised fully. This might explain why initial trials investigating fulvestrant at the 250 mg dose showed only comparable efficacy to
anastrozole or tamoxifen.18,19 The open-label, randomised, phase III
Fulvestrant and Anastrozole Combination Therapy (FACT) trial found
no clinical advantage with the combination of fulvestrant 250 mg plus
anastrozole versus anastrozole alone.21 In contrast, the Southwest
Oncology Group (SWOG), in another open-label, randomised, phase
III trial, reported results favouring this combination approach over
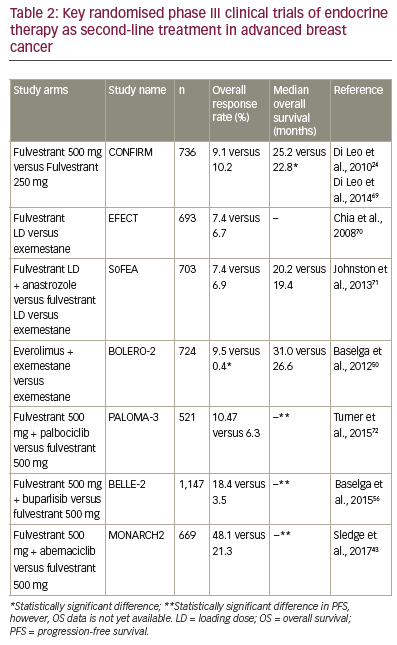
anastrozole alone (Table 2).22 In this study, among women who had not
received prior tamoxifen therapy, the median PFS was 12.6 months with
anastrozole alone versus 17.0 months with fulvestrant plus anastrozole
(hazard ratio, 0.74; 95% confidence interval [CI], 0.59–0.92; p=0.006),
suggesting an increased clinical benefit in patients who were endocrine
therapy-naïve. A potential drug interaction has also been reported with
fulvestrant plus anastrozole, resulting in a decrease in trough anastrozole
concentration in patients in this study.23
Further supporting the effect of fulvestrant dose on efficacy, fulvestrant
500 mg/month versus 250 mg/month was compared in the Comparison
of Faslodex in Recurrent or MBC (CONFIRM), a randomised, doubleblind,
phase III trial.24 Fulvestrant 500 mg was associated with a 19%
reduction in the risk of death and a 4.1 month difference in median
OS compared with fulvestrant 250 mg (Median OS 26.4 months versus
22.3 months, respectively; hazard ratio, 0.81; 95% CI, 0.69–0.96; nominal
p=0.02). Fulvestrant 500 mg regimens therefore offer the possibility of greater antitumour activity than the 250 mg regimen.25,26 Comparison
of the fulvestrant high-dose 500 mg regimen versus anastrozole in the
Fulvestrant fIRst-line Study comparing endocrine Treatments (FIRST) trial
showed a 34% reduction in the risk of progression in patients treated
with fulvestrant (hazard ratio, 0.66; 95% CI, 0.47–0.92; p=0.01).27
To investigate further the potential benefits of fulvestrant 500 mg/
month, and expand upon earlier data suggesting an increased clinical
benefit for fulvestrant in patients who were endocrine therapy naïve,22
the Fulvestrant and AnastrozoLe COmpared in hormonal therapy-
Naïve advanced breast cancer (FALCON) first-line therapy cohort only
randomised, double-blind, multicentre phase III trial was initiated.28 In
this study, there was a statistically significant 21% reduction in the risk
of disease progression or death in women with HR-positive advanced
breast cancer who had been treated with fulvestrant 500 mg (n=230)
compared with those who had received anastrozole 1 mg/day (n=232).
The median PFS was 16.6 months with fulvestrant versus 13.8 months
with anastrozole (hazard ratio, 0.797; 95% CI, 0.637–0.999; p=0.0486).29
Subgroup analysis showed improved PFS in fulvestrant-treated patients
whose disease had not spread to the liver or lungs at baseline, indicating
that fulvestrant would be a particularly advantageous option for patients
with non-visceral disease whereas, for patients with visceral disease,
outcomes were similar.
Enhancing endocrine responsiveness with combination therapies
Despite the development of resistance to endocrine therapies in
some tumours, the oestrogen receptor can still remain functional and
interact with growth factor signalling pathways. Using model systems
of oestrogen receptor-positive breast cancers, resistance to endocrine
therapy has been associated with persistent cyclin D1 expression and
constitutive activation of cyclin-dependent kinase 4 and 6 (CDK4/6).30
Furthermore, treatment with the CDK4/6 inhibitor palbociclib, found
that cell lines representing luminal oestrogen receptor-positive
subtypes were the most sensitive to growth inhibition.31 Preclinical
data have demonstrated growth inhibition by CDK4/6 inhibitors
in HR-positive breast cancer cells.30-32 It has also been demonstrated in
vitro that breast cancer cells can escape hormone dependence due to
hyperactivation of the PI3K pathway, and PI3K or mTOR inhibitors can
block this effect.33 These preclinical data provide a strong rationale for
combining targeted therapies such as CDK4/6, PI3K, or mTOR inhibitors
with endocrine therapy to enhance efficacy (Figure 1).
Cyclin-dependent kinase 4/6 inhibitors
Disordered cell cycle regulation results in uncontrolled cell proliferation
and is therefore an important target for cancer therapies, with the goal of
diverting tumour cells from proliferation, towards a state of non-division.34
One important pathway in cellular proliferation involves CDK4/6, which
coordinate cell cycle progression via reversible combination with cyclin
D.34 It has been demonstrated in vitro that co-targeting the CDK4/6
pathway can restore endocrine sensitivity in cancer cells,31 potentially
improving the efficacy of endocrine therapies in HR-positive breast
cancer patients. Three specific CDK4/6 inhibitors, palbociclib, abemaciclib
and ribociclib are currently being tested in combination with endocrine
therapy in clinical trials.
Palbociclib
Palbociclib is a selective CDK4/6 inhibitor (Figure 1), which prevents
cellular DNA synthesis by preventing downstream phosphorylation of
retinoblastoma protein (Rb) and blocking cell cycle progression from
the G1 to the S phase.31 During preclinical investigation of palbociclib it was found that treatment of cell lines representing luminal oestrogen
receptor-positive subtype were the most sensitive to growth inhibition,
while non-luminal/basal subtypes were the most resistant.31 These
results present a strong rationale for clinical studies of palbociclib and
endocrine therapy combinations in oestrogen receptor-positive breast
cancer patients.31
In the first-line, randomised, phase II PALbociclib: Ongoing trials in
the MAnagement of breast cancer (PALOMA-1) study of patients with
HR-positive MBC, patients treated with palbociclib plus the aromatase
inhibitor, letrozole showed significantly improved PFS (median followup
29.6 months [95% CI 27.9–36.0]) compared with letrozole alone (27.9
months [25.5–31.1]).35 PALOMA-1 included postmenopausal women
with advanced oestrogen receptor-positive and human epidermal
growth factor receptor 2 (HER2)-negative breast cancer who had not
received any systemic treatment (n=165). Median PFS in this patient
subgroup was 20.2 months (13.8–27.5) for those in the palbociclib plus
letrozole group and 10.2 months (95% CI, 5.7–12.6) in the letrozole
monotherapy group (hazard ratio, 0.488; 95% CI, 0.319–0.748; onesided
p=0.0004). On the strength of the data from this trial, the US
Food and Drug Administration granted an accelerated new drug
approval, conditional upon results from PALOMA-2, a randomised,
multicentre, phase III study (n=666) of palbociclib plus letrozole versus
letrozole alone as first-line therapy.36 Survival data are not yet available,
however, PFS was 24.8 months (95% CI, 22.1 to not estimable) in the
palbociclib plus letrozole group, versus 14.5 months (95% CI, 12.9–17.1)
in the placebo plus letrozole group (hazard ratio, 0.58; 95% CI, 0.46–
0.72; p<0.001). The benefit of CDK4/6 inhibition was consistent across
subgroups and unaffected by the presence of ESR1 mutations.
The multicentre, double-blind, randomised phase III PALOMA-3 study
(NCT01942135) (n=521) investigated the combination of fulvestrant plus
palbociclib versus fulvestrant plus placebo as second-line treatment or in
patients with HR-positive, HER2-negative resistant MBC.37 Fulvestrant plus
palbociclib was associated with significant and consistent improvement
in PFS compared with fulvestrant plus placebo, irrespective of the degree
of endocrine resistance, HR expression level, and PIK3CA mutational
status.38 Median PFS was 9.5 months (95% CI, 9.2–11.0) in the fulvestrant
plus palbociclib group and 4.6 months (3.5–5.6) in the fulvestrant plus
placebo group (hazard ratio, 0.46; 95% CI, 0.36–0.59; p<0.0001). Treatment
with palbociclib in combination with fulvestrant was generally safe
and well tolerated, with neutropaenia representing the most common
adverse event. Unlike results seen with chemotherapy, in the palbociclib
arm the rate of febrile neutropaenia was very low (0.9%) despite the
high rate of grade 3–4 neutropaenia (65%). To manage neutropaenia in
patients receiving this drug combination, it is recommended that blood
counts should be monitored before the start of each new cycle as well
as on day 14 of the first two cycles, with appropriate palbociclib dose
delays and reductions.39 Overall, these data show promising efficacy with
fulvestrant plus palbociclib, with manageable adverse events.
Ribociclib
Ribociclib is a small molecule inhibitor of CDK4/6 that exhibits highly
specific inhibitory activity against CDK4/cyclin D1 and CDK6/cyclin D3
complexes.32 In mouse models, ribociclib has shown activity (as a single
agent and in combination with letrozole and PI3K inhibitors), in breast
cancers with intact oestrogen receptor and/or activating aberrations
of PIK3CA/HER-2.32 In the Mammary ONcology Assessment of LEE011’s
(ribociclib’s) efficacy and SAfety (MONALEESA-2) phase III trial, 668
postmenopausal women with HR-positive, HER2-negative advanced
breast cancer, who had not undergone any prior systemic treatment, were randomised to ribociclib plus letrozole, or letrozole plus placebo.40 There
was a 44% improvement in PFS in those who had received ribociclib plus
letrozole, compared with those who had received letrozole alone (hazard
ratio, 0.556; p=0.00000329). At data cut-off, the median PFS was 14.7
months in the letrozole arm, but was not reached in the ribociclib plus
letrozole arm. The observed benefit of CDK4/6 inhibition was consistent
across subgroups. Neutropaenia was the most commonly observed
adverse event, however it was reversible with dose modifications. It
is estimated that results from the MONALEESA-3 trial of ribociclib in
combination with fulvestrant will be available in August 2019.41
Abemaciclib
Abemaciclib is potent inhibitor of Rb phosphorylation and has been
found to inhibit tumour growth in mouse models.42 This compound
has been investigated in combination with endocrine therapy, in two
clinical trials. MONARCH2 (NCT02107703), a study in women with HRpositive,
HER2-negative advanced breast cancer, compared PFS among
patients receiving the CDK4/6 inhibitor, abemaciclib plus fulvestrant
versus fulvestrant alone as second-line treatment or in patients with
resistant disease. Abemaciclib plus fulvestrant significantly extended
PFS compared with fulvestrant alone (median, 16.4 versus 9.3 months;
hazard ratio, 0.553; 95% CI, 0.449 to 0.681; p<0.001).43 Unlike CDK4/6
inhibitors palbociclib and ribociclib, the most common adverse event in
the abemaciclib arm was diarrhoea (86.4%), followed by neutropaenia
(46.0%), nausea (45.1%) and fatigue (39.9%).
MONARCH3 (NCT02246621) is a randomised, double-blind, placebocontrolled,
phase III study investigating anastrozole or letrozole plus
abemaciclib, or placebo in postmenopausal women with HR-positive,
HER2-negative locoregionally recurrent or MBC with no prior systemic
therapy. An interim analysis showed that the combination of abemaciclib
and endocrine therapy significantly prolonged PFS (hazard ratio, 0.543;
p=0.000021).44 Rates of diarrhoea and neutropaenia in the abemaciclib
arm were 81.3% and 41.3%, respectively.
Mammalian target of rapamycin and phosphoinositide 3 kinase inhibition
The PI3K/Akt/mTOR pathway is a prototypic survival pathway that is
constitutively activated in many types of cancer (Figure 1).45 Increased
activation of the PI3K/Akt/mTOR pathway is a common mechanism of
resistance to endocrine therapy,46,47 and therefore inactivation of this
pathway presents an exciting potential target for increasing endocrine
therapy responsiveness.
Mammalian target of rapamycin inhibitors
Temsirolimus is a specific inhibitor of mTOR (Figure 1), which in preclinical
studies was shown to inhibit the proliferation of oestrogen-dependent
breast cancer cell lines.48 In HORIZON (NCT00083993; n=1,112), a phase
III randomised, placebo-controlled study combining temsirolimus
with letrozole as first-line therapy, there was no improvement in PFS
in patients with aromatase inhibitor-naive advanced breast cancer.49
Treatment-emergent adverse events were observed more frequently in
the temsirolimus plus letrozole arm in comparison with letrozole only.
It should be noted that some benefit was seen in exploratory analyses
of the HORIZON trial, where patients aged ≤65 years receiving letrozole
plus temsirolimus showed improved PFS over those receiving letrozole/
placebo, 9.0 versus 5.6 months, respectively (hazard ratio, 0.75; 95% CI,
0.60–0.93; p=0.009).
Everolimus, another specific inhibitor of mTOR (Figure 1), in combination
with the aromatase inhibitor exemestane has been shown to improve
outcomes in patients with MBC resistant to hormone therapies.50 In
the BOLERO-2 (NCT00863655) phase III, randomised trial comparing
everolimus and exemestane, versus exemestane and placebo (n=724)
(Table 2), median PFS was significantly longer in patients receiving
everolimus plus exemestane versus exemestane (4.6-months
prolongation; p<0.0001).51 However, there was no significant
improvement in the secondary endpoint of OS.52 Common adverse
events included stomatitis, anaemia, dyspnoea and hyperglycaemia
were typically of mild or moderate severity, and generally manageable
with dose reduction and interruption. Exploratory analysis from
BOLERO-2 suggests that the efficacy of everolimus is largely
independent of the most commonly altered genes or genetic pathways
in HR-positive, HER2-negative breast cancer.53 The positive results from
BOLERO-2 contrast with those reported for HORIZON and this may, in
part, be a reflection of the different patient populations, in particular, of
the proportion of patients with secondary resistance.
BOLERO-4 (NCT01698918) is the first trial to evaluate the efficacy and
safety of first-line everolimus plus letrozole in postmenopausal women
with oestrogen receptor-positive, HER2-negative metastatic or locally
advanced breast cancer.54 The median PFS was not yet reached at data
cut-off, which was 2 months after the last patient’s first visit. Estimated
PFS rates (95% CI) were 83.6% (77.3–88.2%) and 71.4% (64.0–77.5%) at
6 and 12 months, respectively. The most common adverse events were
stomatitis (67.8%), weight loss (42.6%) and diarrhoea (36.1%). Based on
these results, everolimus combined with letrozole appears to provide clinical benefit in HR-positive, HER2-negative advanced breast cancer
in the first-line setting. However, optimisation of treatment exposure
must be balanced with the adequate management of adverse events.
As such, careful monitoring with appropriate dose reductions and
interruptions is recommended.

Phosphoinositide 3 kinase inhibitors
PI3K CA mutations are frequently found in breast cancer.47 Results with pan
PI3K inhibitors are largely disappointing, showing modest improvement
at best and substantial toxicity. The phase II FERGI (NCT01437566) study
found that the addition of the PI3K inhibitor pictilisib to fulvestrant did
not significantly improve PFS. However, toxicity issues limited pictilisib
dosing, thereby potentially limiting its efficacy.55 BELLE-2 (NCT01610284)
(n=1,147) was a randomised, phase III clinical trial designed to assess the
efficacy of the PI3K inhibitor, buparlisib, plus fulvestrant in breast cancer
patients whose tumours no longer respond to aromatase inhibitors.56
A modest benefit in terms of PFS was observed, with a median PFS of
6.9 months with the combination versus 5.0 months with fulvestrant
alone (hazard ratio, 0.78; p<0.001). The safety profile of the combination
was characterised by transaminitis, hyperglycaemia, rash and mood
disorders, in particular, depression (26.2% of patients with buparlisib plus
fulvestrant versus 8.9% with fulvestrant alone).
Patients progressing after mTOR inhibition may still receive benefit from
re-targeting the same pathway, although only in those with PIK3CA
mutation. In BELLE-3 (NCT01633060), patients had HR-positive, HER2-
negative, aromatase inhibitor-treated, locally advanced or MBC that
had either progressed on or after treatment with everolimus. In total,
432 patients were randomly assigned (2:1) to receive daily buparlisib
plus fulvestrant or placebo plus fulvestrant.57 Median PFS for patients in
the buparlisib arm was 3.9 months compared with 1.8 months for those
in the placebo arm (p<0.001). Among patients with PIK3CA mutations,
PFS was 4.7 months for those in the buparlisib arm compared with 1.4
months for the placebo arm (p<0.001).
In summary, PI3K/mTOR pathway inhibition has shown substantial
benefit in resistant disease, especially with mTOR inhibitors; it may be
that these agents are best used in acquired resistance (as in BOLERO-2),
whereas in sensitive disease (e.g. HORIZON) the benefit is less clear.
Future development is focusing on alpha-selective (beta-sparing)
inhibitors. Since side-effects are often mediated through beta effects,
the hope is for better tolerability, allowing improved targeting and higher efficacy. Further results from phase III programmes are expected in 2018
and 2019 (Table 3).
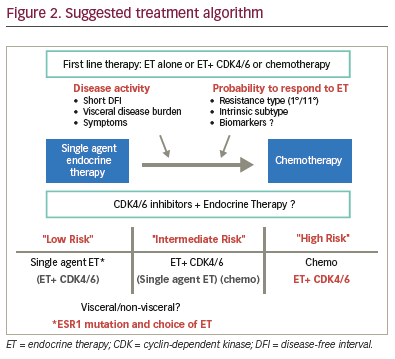
Clinical decisions for treatment strategy
Where clinicians previously only had the options for chemotherapy with
single-agent endocrine therapies, multiple strategies are now available.
It is to be determined whether all patients suitable for endocrine therapy
should receive combination therapy or whether there remains a role for
single-agent endocrine therapy. Single-agent endocrine therapy shows
consistent activity with PFS of 12–14 months for aromatase inhibitors
and 16 months for fulvestrant 500 mg, establishing fulvestrant 500 mg
as the most effective single-agent endocrine therapy. The CDK4/6
combinations seem to lead to substantially better PFS than aromatase
inhibitors. However, currently no advantage has been shown on OS,
and combination strategies increase the risk of side effects and are
also more expensive. Even if first-line trials had shown benefit in terms
of OS, it would not answer the question as to whether similar effects
can be achieved with cross-over. CDK4/6 inhibitors show comparable
overall benefits relative to aromatase inhibitors and fulvestrant 500
mg in second-line therapy. Therefore, it is reasonable to still consider
single-agent endocrine therapy for some patients, ideally those at low
risk with low activity and presumed sensitivity (Figure 2). This choice
depends on a range of factors, such as: the type of adjuvant treatment
received, length of disease-free interval, organ function, disease extent
at recurrence, pre/postmenopausal status, adverse effect profile
of therapy, and tumour biology (i.e., HR-receptor expression, HER2
expression levels, and mutational status). Unfortunately, there is a
great deal of heterogeneity in HR-positive MBC and so there is a critical
need for the development of predictive biomarkers to allow improved
guidance in treatment choice.
When recommending appropriate endocrine therapy, fulvestrant’s
efficacy must be weighed against its intramuscular administration, which
necessitates more frequent visits. Particular benefit with fulvestrant
has been seen in those with non-visceral disease and ESR1 mutation
status. Impressive response rates have been reported with CDK4/6
combinations, with objective response rates in excess of 40%. This is
in the range of chemotherapy response rates in phase III trials with
endocrine receptor-positive disease. Combination strategies might
therefore allow more patients to benefit from endocrine therapy rather
than chemotherapy initially, even those patients with higher risk disease.
Guidance on selection and sequencing of treatments should be reexamined
following the availability of survival data, both OS and PFS,
from the ongoing FALCON, PALOMA-2 and MONALEESA-2 trials. These
three trials included different patient groups, with presumably different
endocrine sensitivity. However, the efficacy of anastrozole appears
consistent across the trials, a finding that has not yet been explained.
In phase III trials, the response rates observed with CDK4/6 inhibitor
combination have been similar to that for chemotherapy. Based on
current available data, recent guidelines from the American Society for
Clinical Oncology (ASCO) made the following key recommendations for
endocrine therapy in HR-positive MBC39
- Patients with tumours and any level of HR expression should be
offered hormonal therapy. - Therapy recommendations should take into account the type of
adjuvant treatment, disease-free interval, and extent of disease at the
time of recurrence. If recurrence occurs >12 months following prior
therapy, a specific endocrine therapy may be reused. - With the exception of patients with life-threatening disease, or
those on adjuvant endocrine therapy who experience rapid visceral
recurrence, endocrine therapy should be recommended as the initial
treatment option. - Treatment should be continued until disease progression occurs.
- Combined use of endocrine therapy and chemotherapy is not
recommended. - All patients, including those receiving first-line treatment should be
encouraged to consider enrolling in clinical trials.
The choice of single-agent endocrine therapy can be influenced by
biology (ESR1 mutational status) and disease pattern (e.g. non-visceral versus visceral). In light of the assumed impact of ESR1 mutations on
the outcome of patients in response to endocrine therapy, detecting
ESR1 mutations is likely to be of interest to individualise treatment
of MBC.5 ESR1-status does not appear to determine response to
selective oestrogen receptor downregulators.58,59 However, preclinical
studies revealed relative resistance of the mutations to tamoxifen and
fulvestrant but effective inhibition with high doses.60-64 Retrospective
analyses of ESR1 mutations in baseline plasma circulating tumour
DNA from completed clinical trials suggest that these mutations are
prognostic and predictive of resistance to aromatase inhibitors in
metastatic disease. However, we need prospective studies to confirm
these results and to determine the best treatment combinations for
patients with ESR1 mutations. Clinical development of novel agents to
overcome resistance engendered by ESR1 mutations is also needed.
Higher doses of fulvestrant appear to improve outcomes for patients
with these mutations.59
Concluding remarks
Patients with low disease burden, slow progression and presumed
endocrine sensitivity might still be considered for single-agent
endocrine therapy, whereas patients with more aggressive disease
e.g. visceral metastases, could benefit from combination therapy.
There is an apparent improvement in efficacy of fulvestrant in
the patient group with non-visceral versus visceral disease in the
FALCON trial (Figure 2), and similar observations were reported in the
FIRST65 and CONFIRM24 trials. Current guidelines still recommend the
use of endocrine therapy for visceral patients not in visceral crisis,
however.2,4 Overall, there is some evidence in support of a more
tailored approach, although the ‘one-size fits-all’ tactic cannot be
dismissed at present.