Colorectal cancer (CRC) is a commonly diagnosed malignancy and the second-leading cause of cancer death in the USA, with 147,950 estimated new cases and 53,200 estimated deaths in 2020.1 Despite CRC screening, approximately 20% of patients are diagnosed with metastatic CRC (mCRC), which carries a 14.2% 5-year survival rate.2
Over the last two decades, mCRC treatment has become more personalized, with a better understanding of tumor heterogeneity, including microsatellite instability (MSI) and mutations in genes such as KRAS, NRAS, and BRAF. The availability of molecularly tailored treatments has reshaped the therapeutic landscape for many patients with mCRC. Approximately 8–12% of mCRCs contain a mutation in BRAF, also known as v-raf murine sarcoma viral oncogene homolog B.3–6 BRAF is a signal transduction protein involved in the mitogen-activated protein kinase (MAPK) pathway.3 Here we review the prognostic and therapeutic implications of BRAF-mutated CRC.
BRAF pathway and mutations in malignancies
The BRAF gene is located on chromosome 7 (7q34). It encodes the BRAF protein, a serine/threonine protein kinase, which plays a role in the regulatory MAPK/extracellular signal-regulated kinase (ERK) signaling pathway during cell growth, proliferation, differentiation, and apoptosis.7,8 Mutations in BRAF result in downstream phosphorylation of mitogen-activated extracellular signal-regulated kinase (MEK) and ERK, leading to activation of the MAPK pathways. This oncogenic mutation stimulates cell proliferation and metastasis (Figure 1).
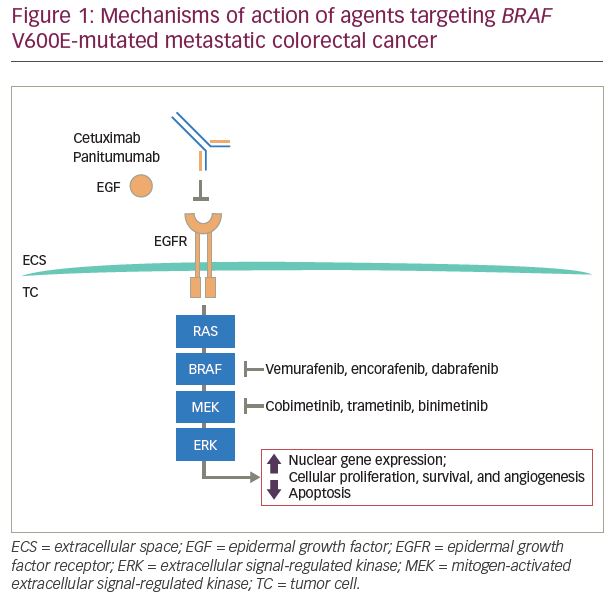
BRAF mutations have been identified with varying incidence in multiple malignancies, including melanoma, CRC, hairy cell leukemia, lung cancer, ovarian cancer, Langerhans cell histiocytosis, and papillary thyroid carcinoma.9–14 The BRAF inhibitors, dabrafenib, encorafenib, and vemurafenib, are US Food and Drug Administration (FDA)-approved therapies for patients with BRAF-mutated melanoma. Median progression-free survival (PFS) of patients with melanoma treated with these inhibitors ranges from 5–10 months.15–18
BRAF mutations in colorectal cancer
Most BRAF mutations in CRC appear in primary tumors located in the proximal colon and first two-thirds of the transverse colon. BRAF-mutated tumors tend to be larger than average, are associated with serrated polyp morphology, and have higher rates of distant metastases.19 Eighty percent of hyperplastic microvascular polyps harbor BRAF mutations, and these polyps present in the proximal colon as sessile serrated adenomas.19 These types of polyps have high malignant potential and are under-detected by conventional colonoscopies, leading to their ability to progress without being detected.19 BRAF-mutated CRCs metastasize readily to the peritoneum and distant lymph nodes, but less readily to the lungs. BRAF mutations occur more commonly in mCRCs that are microsatellite instable. Women and individuals over the age of 65 years are more likely to have mCRCs that harbor mutated BRAF.5,20
In response to the binding of extracellular growth factors, receptor tyrosine kinases activate RAS, which activates and induces the formation of RAF dimers to propagate and transduce downstream signaling to promote cell proliferation and differentiation. Mutations in BRAF fall into one of three categories based on the biochemical and signaling properties of their encoded proteins. Class 1 contains the most common BRAF mutation known as BRAF V600E, which is the product of a thymine to adenine base transversion at codon 600 of exon 15 of BRAF. This mutation, which leads to a substitution of valine for glutamate at position 600 of the encoded protein, is observed in approximately 10% of all patients with mCRC and accounts for around 80% of all BRAF mutations.21 The resulting aberrant protein exhibits high kinase activity and can signal independently of RAS activation.21 BRAF mutations in classes 2 and 3 occur at different locations in the BRAF gene, encoding non-V600E mutated proteins. These non-V600E BRAF mutations are observed in mCRCs of approximately 2% of patients and account for around 20% of all BRAF mutations (about 10% in each of class 2 and 3).21 Class 2 mutations, like class 1, give rise to proteins that signal independently of RAS. In contrast, class 3 BRAF mutations encode proteins that have enhanced binding to RAS and CRAF and lead to RAS-dependent signaling.6
The activities of these different classes of BRAF mutations correlate with differences in the clinical characteristics of BRAF-mutant mCRC and response to targeted therapy. Tumors with class 1 BRAF mutations (BRAF V600E-mutant) are typically found in right-sided, high-grade, MSI-high tumors and are associated with a worse patient prognosis compared with BRAF-wild type (WT) tumors. Patients with BRAF V600E-mutated mCRC have a median overall survival of 4–6 months after failure of initial therapy, compared with 11–14 months for all patients with mCRC.22,23 Class 2 and 3 (BRAF non-V600E mutant) tumors have been observed in younger patients with left-sided tumors, and the prognosis for these patients is similar to those with BRAF-WT tumors.6
The class of BRAF mutation is clinically relevant because it has been shown to indicate prognosis, as well as effectiveness of the BRAF inhibitors, vemurafenib, dabrafenib, and encorafenib, and the anti-epidermal growth factor receptor (EGFR) monoclonal antibodies, cetuximab and panitumumab.22,23 Upon further examination of clinical characteristics of CRC tumors with non-V600E BRAF mutations, patients with RAS-WT tumors and class 2 BRAF-mutated CRC had shorter survival compared with those with RAS-WT and class 3 BRAF-mutant CRC.25 EGFR is a receptor tyrosine kinase in the MAPK pathway; RAS and BRAF operate downstream of EGFR.6 While EGFR inhibitors improve overall survival in CRC, CRC tumors with activating RAS mutations do not benefit from treatment with anti-EGFR antibodies.24 Because class 1 BRAF mutations are RAS activating, anti-EGFR antibody monotherapy is ineffective. Similarly, class 2 BRAF-mutant CRCs rarely respond to EGFR therapy, suggesting that class 2 BRAF mutations confer resistance. 25 Of 36 patients with non-V600E BRAF mutations, patients with class 2 and 3 BRAF mutations did not have any objective response to anti-EGFR monotherapy.26 Class 3 mutated CRCs that did not respond to anti-EGFR therapy have been shown to be linked to underlying activation of other receptor tyrosine kinases leading to RAS activation.25 However, considering that a large portion of patients with class 3 BRAF mutations responded to anti-EGFR therapy, the main driver in these tumors is thought to be EGFR activation. These patients likely have tumors that are sensitive to EGFR inhibition.26
Treatment in BRAF V600E-mutated CRC
Patients with BRAF V600E-mutated CRC currently have shorter PFS on chemotherapy, an increased rate of disease recurrence after surgical resection, and poorer overall survival.20,27 As mentioned above, BRAF mutations in CRC confer resistance to anti-EGFR therapy,28–32 and tend to be refractory to standard chemotherapy.33 Upfront intensive chemotherapy is often used, based on the phase III TRIBE trial results (ClinicalTrials.gov identifier: NCT00719797), which observed benefit of fluorouracil, leucovorin, oxaliplatin, and irinotecan (FOLFOXIRI)/bevacizumab over fluorouracil, leucovorin, and irinotecan (FOLFIRI)/bevacizumab in patients with
BRAF-mutated mCRC.34 In the TRIBE2 trial (ClinicalTrials.gov identifier: NCT02339116), which included 10% BRAF-mutated patients in each arm, the combination of FOLFOXIRI/bevacizumab improved PFS during both upfront and pre-planned re-introduction after disease progression, as well as longer overall survival (27.6 versus 22.6 months, hazard ratio [HR] 0.81, 95% confidence interval [CI] 0.67–0.98, p=0.033).35
Although the BRAF V600E mutation has been identified and successfully targeted in multiple other advanced cancers, BRAF-inhibitor monotherapy shows limited efficacy in cases of BRAF V600E-mutated mCRC. Vemurafenib is a small tyrosine kinase inhibitor that specifically targets the adenosine triphosphate-binding domain of BRAF V600E, and has been shown to decrease activation of the MAPK pathway in advanced malignancies with BRAF mutations.4 In mCRC, vemurafenib monotherapy is rarely effective at reducing tumor growth. This decreased efficacy is attributed to incomplete inhibition of MAPK signaling and a reflexive activation of EGFR, which promotes tumor progression through alternative pathways, bypassing BRAF.4 The combination of vemurafenib plus trametinib (a MEK inhibitor) resulted in response rates as high as 76% in BRAF V600E-mutated metastatic melanoma. In contrast, this same combination yielded only a 12% response rate in BRAF V600E-mutated CRC.36 In a phase IB study of 17 patients with BRAF V600E-mutated mCRC, the addition of vemurafenib to a combination of irinotecan plus cetuximab yielded a 35% response rate and a median PFS of 7.7 months.4 Patients with radiographically observed responses and stable disease also exhibited reductions in the percentage of BRAF V600E cell-free DNA.4
This regimen was further studied in the phase II SWOG S1406 study (ClinicalTrials.gov identifier: NCT02164916), which randomized patients with previously treated BRAF V600E-mutated mCRC 1:1 to irinotecan plus cetuximab versus irinotecan plus cetuximab and vemurafenib. The primary endpoint of PFS was significantly longer in the arm that received vemurafenib: 4.3 versus 2.0 months (HR 0.48, 95% CI 0.31–0.75; p=0.001).22
In addition to combined inhibition of EGFR and BRAF, the inhibition of the MAPK pathway has been explored through the combined inhibition of the MEK, EGFR, and BRAF. The phase III BEACON study (ClinicalTrials.gov identifier: NCT02928224) randomized patients with previously treated BRAF V600E-mutated mCRC 1:1:1 to triplet therapy with encorafenib (BRAF inhibitor), cetuximab (anti-EGFR antibody), and binimetinib (MEK inhibitor); doublet therapy with encorafenib and cetuximab; or the control arm of chemotherapy (FOLFIRI or irinotecan) with cetuximab (N=665).37 Patients treated with the triplet and doublet therapy had significantly longer median overall survival when compared with the control arm: 9.3 and 9.3 versus 5.9 months, respectively. The median overall response rate was also higher with the triplet (27%) and doublet (20%) therapy compared with the control arm of chemotherapy (2%, p<0.0001).37 As there was not a significant difference in overall survival between the triplet and doublet therapies, doublet therapy with encorafenib and cetuximab received FDA approval for patients with BRAF V600E-mutated mCRC after prior therapy on April 8, 2020.38 This study’s quality-of-life assessment revealed the doublet and triplet regimens were not different, but both regimens provided improved patient-reported quality of life compared with the control arm.39 The ongoing phase III ANCHOR trial is evaluating the triplet therapy in the
first-line setting (ClinicalTrials.gov Identifier: NCT03693170).
Future approaches to treating BRAF-mutated CRC
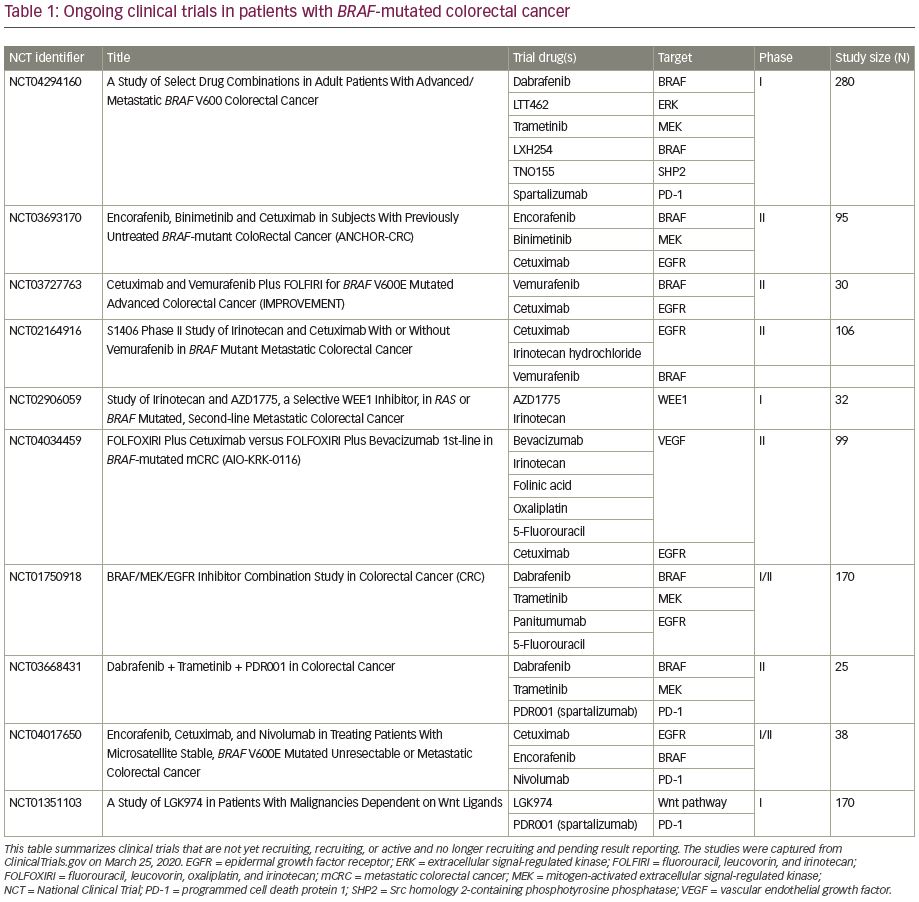
While BRAF V600E mutations are currently a poor prognostic marker, the use of targeted therapies as well as high prevalence of MSI-high, which lends potential benefit from checkpoint inhibitors, are promising therapeutic avenues. Future clinical trials are accruing patients with BRAF-mutated CRC to test multi-agent combinations that inhibit various signaling proteins along the MAPK pathway. Novel targeted agents include ERK inhibitors, e.g., LTT462; BRAF inhibitors, e.g., LXH254; Src homology 2-containing phosphotyrosine phosphatase (SHP2) inhibitors, e.g., TNO155; WEE1 kinase inhibitors, e.g., AZD1775; and Wnt inhibitors, e.g., LGK974 (Table 1). These novel agents are being combined together, added to traditional chemotherapy as well as immunotherapy, in the hope of overcoming the treatment resistance conferred by BRAF mutations. One phase Ib clinical trial is utilizing combinations of targeted agents in the hope to prove the safety of combinations, as well as overcoming potential downstream resistance mechanisms (ClinicalTrials.gov identifier: NCT04294160). Novel combinations include a doublet BRAF inhibitor with ERK inhibitor; triplet arms that include a BRAF inhibitor, ERK inhibitor, and MEK inhibitor; and targeted agents plus immunotherapy or chemotherapy to review safety as well as efficacy. The utilization of concomitant MSI and BRAF mutation testing may help predict response to checkpoint inhibition. Triplet combinations of BRAF inhibition, MEK inhibition, and programmed cell death protein 1 (PD-1) inhibition have been tested on BRAF-mutated melanomas in the phase III COMBI-i trial with acceptable safety profiles and promising response rates.40 This same combination is being testing in BRAF-mutated CRC (ClinicalTrials.gov identifier: NCT03668431). Given its recent FDA approval, encorafenib and cetuximab will likely form the backbone of future studies combining anti-BRAF/EGFR therapy with chemotherapy and other targeted agents.
Radiological studies have also explored the use of imaging factors to predict which patients are more likely to have a BRAF mutation in their tumors. One study (N=155) showed BRAF mutations were more common in female patients (p=0.007), older patients (p=0.001), and right-sided tumors (p=0.001).38 Radiologically significant factors seen in BRAF-mutated tumors, as opposed to BRAF-WT tumors, included right-sidedness (p=0.002); heterogeneous enhancement, possibly due to association between BRAF and a mucinous histology (p=0.039); and lack of non-peritoneal metastases (p=0.043).41
In addition to adenocarcinoma CRC, BRAF V600E mutations have recently been identified in colon neuroendocrine tumors at rates as high as 28% and may benefit from targeted therapy as well.35 This population has unique and complex biological differences compared with BRAF-mutated CRC adenocarcinoma as well as melanoma and will need to be tested in basket trials.
Conclusion
BRAF mutations in mCRC have traditionally portended a poor prognosis and decreased benefit from standard therapies. Although recent studies have shown improved overall survival and response rates with combined encorafenib and cetuximab, there is still a need to identify novel therapy combinations that can further extend survival outcomes of BRAF-mutated patient populations.